


What Is Hemoglobin Made Of?
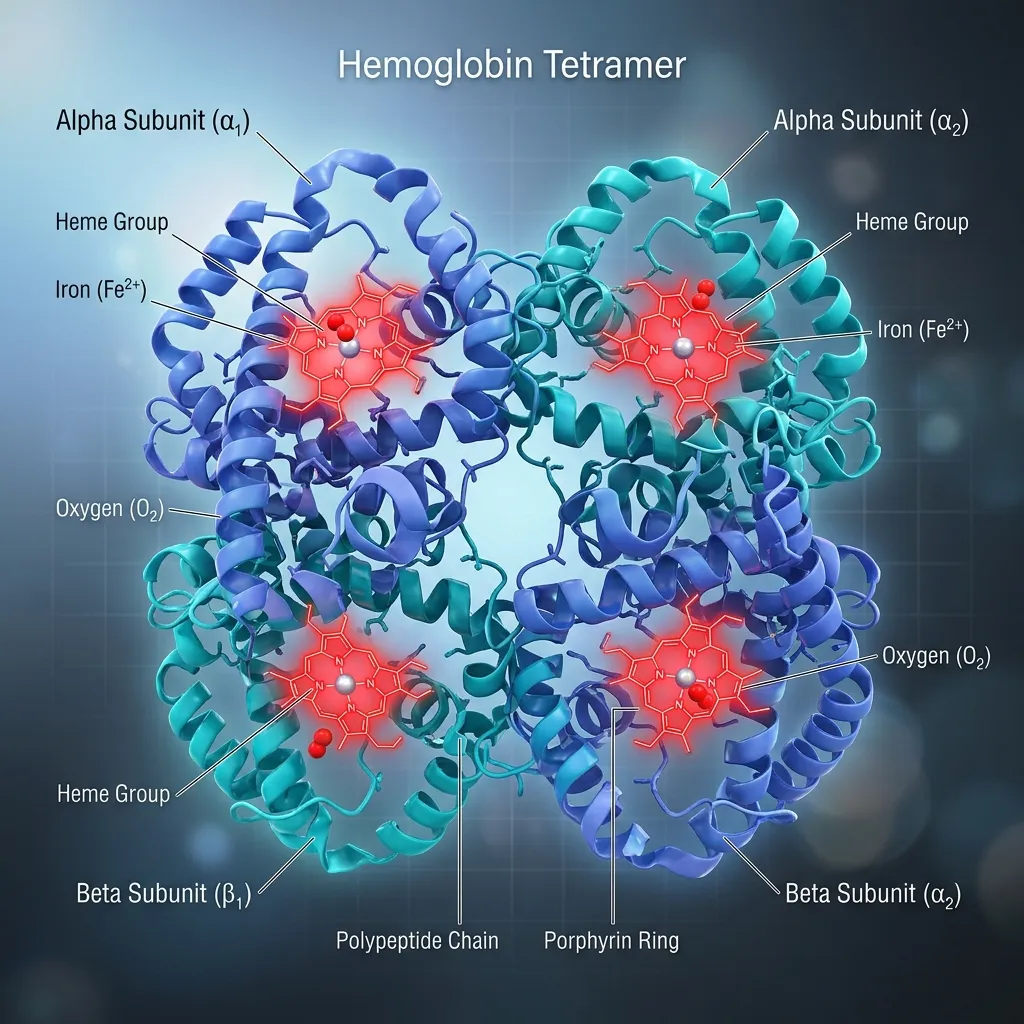 Hemoglobin is the oxygen-carrying protein inside red blood cells. Its main job is simple to state but vital: pick up oxygen in the lungs and deliver it to every tissue in the body. It also helps carry carbon dioxide back to the lungs and supports blood pH balance.
Hemoglobin is the oxygen-carrying protein inside red blood cells. Its main job is simple to state but vital: pick up oxygen in the lungs and deliver it to every tissue in the body. It also helps carry carbon dioxide back to the lungs and supports blood pH balance.
Each hemoglobin molecule has a four-part design known as a quaternary structure. It contains four globin chains and four heme groups. Adult hemoglobin (HbA) is made of two alpha chains and two beta chains, locked together into one functional unit.
The Globin Chains: A Closer Look
The two adult globin chains may look similar, but they differ in key ways.
Alpha globin chain characteristics:
- Contains 141 amino acids
- Produced by genes on chromosome 16
- Humans carry four alpha globin genes (two from each parent)
Beta globin chain characteristics:
- Contains 146 amino acids
- Produced by the HBB gene on chromosome 11
- Humans carry two beta globin genes (one from each parent)
This genetic setup is one of the most important parts of the alpha and beta globin comparison. Because there are four alpha genes but only two beta genes, the two chains follow different inheritance patterns and respond differently to genetic mutations.
The Heme Group: The Oxygen Carrier
Tucked inside each globin chain sits a heme group. Each heme group holds a single iron atom at its center, and this iron is the exact spot where oxygen attaches.
Because every hemoglobin molecule has four heme groups, it can carry up to four oxygen molecules at once. The iron must stay in its ferrous (Fe²⁺) state to bind oxygen properly. If it oxidizes, it can no longer do its job. For a deeper look at this process, see our guide on hemoglobin structure and oxygen binding.
Alpha vs Beta Globin Function: The Core Differences
Now we reach the heart of the matter. The alpha vs beta globin function difference becomes clearest when you look at how these chains handle oxygen, how they hold the molecule together, and when the body produces them.
How Do Alpha and Beta Globin Chains Bind Oxygen?
On their own, individual globin chains can still bind oxygen. An isolated alpha or beta chain will grab an oxygen molecule readily. But here is the catch: a single chain holds oxygen too tightly to be useful. It would pick up oxygen in the lungs and then refuse to let go in the tissues.
The magic happens when the chains work as a team. In the assembled tetramer, alpha and beta chains communicate during oxygen binding. When one chain grabs an oxygen molecule, it slightly changes the shape of the whole protein, making it easier for the next chain to bind. This is called cooperative binding, and it is a true team effort between alpha and beta chains.
This cooperation depends on two shapes:
- Tense (T) state: Low oxygen affinity, common in the tissues where oxygen is released.
- Relaxed (R) state: High oxygen affinity, common in the lungs where oxygen is loaded.
As oxygen binds, hemoglobin shifts from the T state toward the R state. This switching is an allosteric effect, meaning activity at one site changes activity at another. The Bohr effect fine-tunes the process further: in active, acidic tissues with high carbon dioxide, hemoglobin releases more oxygen exactly where cells need it most. Neither chain can do this alone—it takes both alpha and beta chains working together.
How Do Alpha and Beta Chains Hold Hemoglobin Together?
Beyond oxygen handling, the two chains play a structural role. The places where alpha and beta chains touch are called alpha-beta interfaces, and they are key contact points that hold the molecule together.
These interfaces do more than glue the chains in place. They are where the chains pass signals during cooperative binding. When the protein shifts between the T and R states, the alpha-beta interfaces slide and rotate slightly. This movement is part of what allows the chains to coordinate their oxygen handling.
In short, alpha and beta chains keep each other stable. Take one away, and the partner becomes unstable and prone to clumping—a problem we will return to when discussing thalassemia.
When Does the Body Make Alpha and Beta Chains?
One striking part of the difference between alpha and beta globin chains is timing. The body does not produce these chains in the same way across a lifespan.
Before birth, fetal hemoglobin (HbF) dominates. HbF is made of two alpha chains and two gamma chains, not beta. The gamma chains give fetal hemoglobin a higher oxygen affinity, letting the fetus pull oxygen across the placenta from the mother’s blood.
After birth, the body shifts to adult hemoglobin (HbA), made of two alpha chains and two beta chains. Notice the pattern: alpha chains are present both before and after birth, while beta chains only take over after birth. Alpha globin is the constant partner; beta globin is the adult specialist.
This handoff is called globin gene switching, and it is mostly complete by six months of age. It is controlled by transcription factors and epigenetic signals. To understand how this transition works, read our guide on the globin gene switching mechanism.
How Do Alpha and Beta Globin Defects Cause Different Diseases?
Because alpha and beta chains differ in their genes and behavior, defects in each cause different disorders. This is one of the most clinically important parts of the alpha and beta globin comparison.
Alpha Thalassemia
Alpha thalassemia happens when one or more of the four alpha globin genes are deleted or mutated.
The severity depends on how many genes are affected:
- One gene affected: silent carrier, usually no symptoms.
- Two genes affected: alpha thalassemia trait with mild anemia.
- Three genes affected: Hemoglobin H disease, with moderate to severe anemia.
- Four genes affected: Hb Barts and hydrops fetalis, a life-threatening condition before birth.
When alpha chains run short, excess beta chains build up and form abnormal groups like Hemoglobin H.
Beta Thalassemia
Beta thalassemia results from mutations in the beta globin genes, which reduce or stop beta chain production.
It comes in several forms:
- Thalassemia minor (trait): mild, often symptomless.
- Thalassemia intermedia: moderate symptoms.
- Thalassemia major: severe, transfusion-dependent anemia.
When beta chains run short, excess alpha chains pile up. These free alpha chains are highly unstable and toxic, which is why beta thalassemia is often more severe than many forms of alpha thalassemia. You can explore this further in our guide on alpha and beta globin chain imbalance.
What Happens When Alpha and Beta Globin Don’t Align?
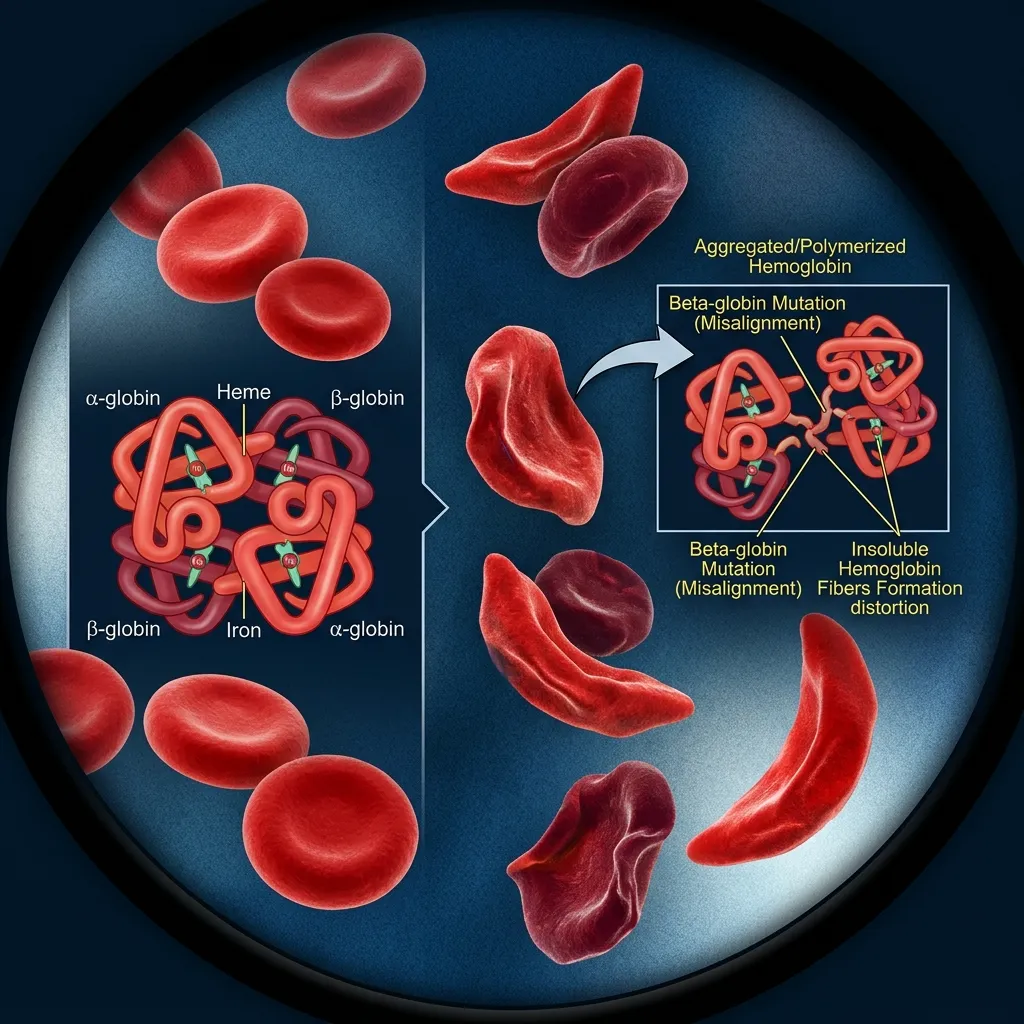 The whole system depends on balance. The body normally makes alpha and beta chains in nearly equal amounts. When that balance breaks, the consequences ripple through the red blood cells.
The whole system depends on balance. The body normally makes alpha and beta chains in nearly equal amounts. When that balance breaks, the consequences ripple through the red blood cells.
Why Are Unpaired Globin Chains So Harmful?
Unpaired chains are the core problem in thalassemia. A chain without a partner cannot form normal hemoglobin. Instead, it clumps together inside the developing red blood cell.
These clumps are toxic. They damage cell membranes, trigger oxidative stress, and mark the cell for early destruction. Excess alpha chains (seen in beta thalassemia) are especially damaging because they are so unstable. Excess beta chains (seen in alpha thalassemia) are generally less toxic, which partly explains the difference in disease severity.
How Does Globin Imbalance Lead to Anemia?
When damaged red blood cells die in the bone marrow before they mature, the result is ineffective erythropoiesis. The bone marrow works overtime to make new cells, but most are destroyed before reaching the bloodstream.
This vicious cycle leads to chronic anemia, bone marrow expansion, skeletal changes, and enlarged organs like the spleen and liver. It also boosts iron absorption, which can cause iron overload over time. For a detailed look at this process, see our guide on ineffective erythropoiesis.
How Are Globin Disorders Diagnosed and Treated?
Doctors use several tools to diagnose globin chain disorders:
- Complete blood count (CBC): Often shows small, pale red blood cells.
- Hemoglobin electrophoresis: Separates hemoglobin types to distinguish alpha from beta thalassemia.
- Genetic testing: Confirms specific mutations and carrier status.
Treatment depends on severity. Options include:
- Blood transfusions to maintain healthy hemoglobin levels.
- Iron chelation therapy to remove excess iron from transfusions.
- Bone marrow or stem cell transplant, the only established cure for severe cases.
- Gene therapy, an emerging approach that corrects or reactivates globin genes. Some newer therapies use CRISPR to switch fetal hemoglobin back on, helping restore balance.
Beyond Oxygen Transport: Other Globin Functions
Carrying oxygen is the headline job, but globin chains do more. Hemoglobin also interacts with nitric oxide (NO), a signaling molecule that helps control blood vessel width and blood flow. By binding and releasing NO, hemoglobin plays a part in regulating circulation.
Hemoglobin also interacts with other proteins and molecules inside the red blood cell, including 2,3-BPG, which adjusts how tightly the protein holds oxygen. These extra roles remind us that alpha and beta chains are not just oxygen taxis—they are active players in several body systems.
How Globin Chain Balance Maintains Healthy Red Blood Cells
A healthy red blood cell depends on a precise balance between alpha and beta globin production. The body carefully regulates globin gene expression so that the correct number of chains is available to build hemoglobin molecules. When production remains balanced, hemoglobin forms efficiently, oxygen transport works properly, and red blood cells survive their normal lifespan.
Problems arise when one type of globin chain is produced in excess while the other is lacking. Unpaired globin chains can become unstable and accumulate inside developing red blood cells. These excess chains create oxidative stress, damage cell membranes, and interfere with normal cell maturation. As a result, many red blood cells are destroyed before they ever enter circulation.
This imbalance is the underlying cause of disorders such as alpha and beta thalassemia. The severity of symptoms often depends on how large the imbalance becomes. Understanding globin chain balance helps explain why even small genetic changes can have major effects on blood health and oxygen delivery.
Future Research and Therapeutic Advances in Globin Disorders

The alpha vs beta globin function difference comes down to genetics, structure, timing, and disease. This alpha and beta globin comparison shows that alpha globin chains have 141 amino acids and come from chromosome 16, with four genes per person. Beta globin chains have 146 amino acids and come from chromosome 11, with two genes per person. Alpha is the constant partner present before and after birth, while beta is the adult specialist.
Yet despite these differences, the difference between alpha and beta globin chains does not change their shared purpose. The two chains depend on each other completely. Their cooperation makes efficient oxygen transport possible, and their balance keeps red blood cells healthy. When that balance tips, thalassemia and related disorders follow.
If you or a family member may be affected by a hemoglobin disorder, the best next step is proper diagnosis through clinical and genetic testing. Speak with a hematology specialist about your options, and explore our related guides on types of thalassemia: alpha vs beta, alpha globin vs beta globin function in hemoglobin, and hemoglobin synthesis disorders to keep learning.