


What Is Globin Gene Switching and Why Does It Matter?
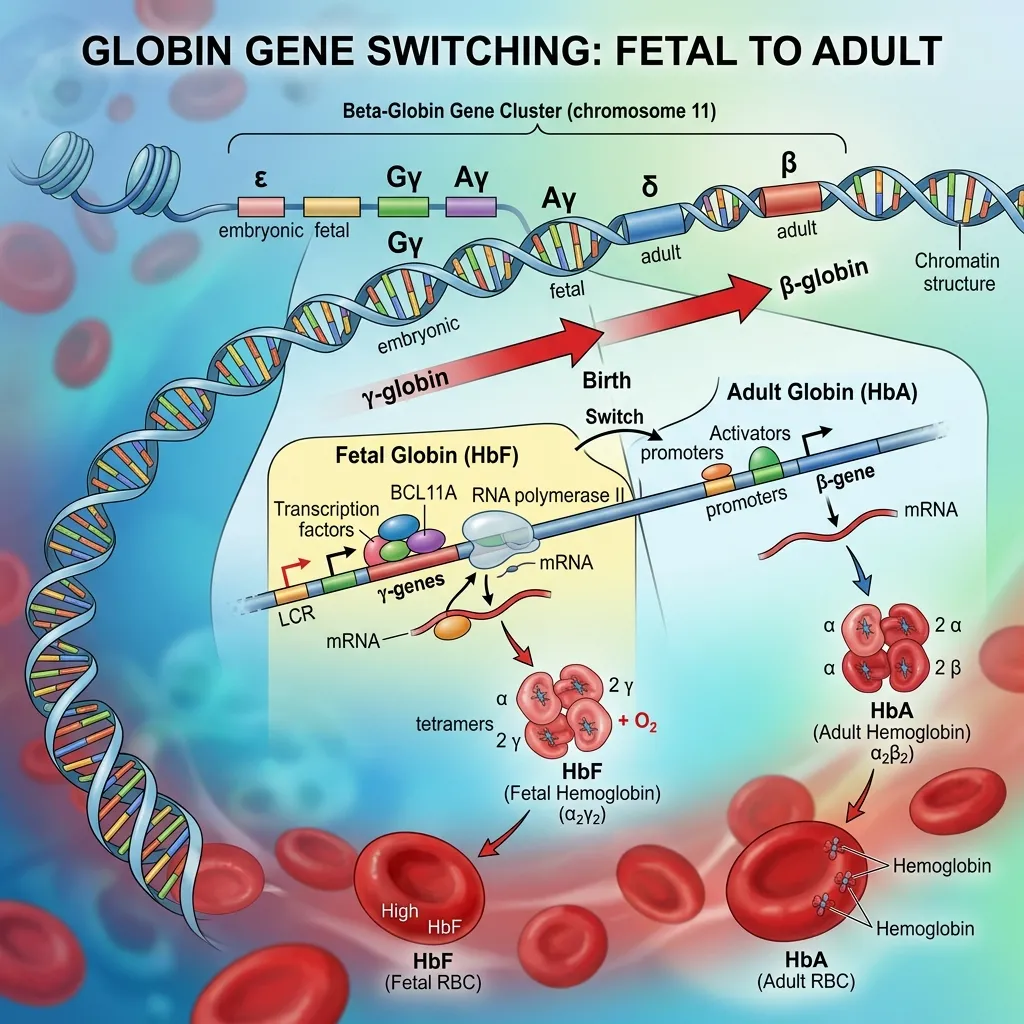 Globin gene switching is the regulated process by which the body changes which globin genes it expresses at different stages of life. Hemoglobin, the oxygen-carrying protein inside red blood cells, is built from four globin chains. The exact chains used depend on whether a person is an embryo, a fetus, or an adult.
Globin gene switching is the regulated process by which the body changes which globin genes it expresses at different stages of life. Hemoglobin, the oxygen-carrying protein inside red blood cells, is built from four globin chains. The exact chains used depend on whether a person is an embryo, a fetus, or an adult.
Three main types of hemoglobin appear across human development:
- Fetal hemoglobin (HbF): Made of two alpha chains and two gamma chains, it dominates before birth and binds oxygen tightly to draw it from maternal blood.
- Adult hemoglobin (HbA): Made of two alpha chains and two beta chains, this is the primary hemoglobin after infancy and accounts for roughly 97 percent of adult hemoglobin.
- Hemoglobin A2 (HbA2): A minor adult hemoglobin made of two alpha chains and two delta chains, usually making up less than 3.5 percent of total hemoglobin.
The central event is the fetal-to-adult hemoglobin switching process, where gamma-globin production fades and beta-globin production rises. This switch begins around birth and is mostly complete by six months of age. Understanding it matters because the timing and control of this switch directly affect the severity of inherited blood disorders.
What Molecular Machinery Controls Hemoglobin Gene Regulation?
The hemoglobin gene regulation mechanism explained at the molecular level involves clusters of genes, powerful control regions, and a cast of regulatory proteins working in coordination.
The Alpha and Beta Globin Gene Clusters
Globin genes are organized into two clusters on separate chromosomes. The alpha-globin gene cluster sits on chromosome 16, while the beta-globin gene cluster sits on chromosome 11. The beta cluster contains the genes arranged in the order they switch on during development: epsilon (embryonic), two gamma genes (fetal), then delta and beta (adult). This physical arrangement mirrors the timeline of development.
Key Regulatory Elements: LCR, Promoters, and Enhancers
Several DNA elements direct globin gene activity:
- Locus Control Region (LCR): A master switch located upstream of the beta-globin cluster, the LCR boosts expression of whichever globin gene is active at a given developmental stage.
- Promoters: Located at the start of each gene, promoters serve as docking sites where the cell’s machinery begins reading the gene.
- Enhancers: These DNA sequences increase gene activity and help the LCR connect with the correct gene at the correct time.
Transcription Factors That Drive the Switch
Transcription factors are proteins that bind DNA and turn genes on or off. Two factors stand out in the gamma to beta globin switch in development. BCL11A acts as a powerful silencer of gamma-globin, helping shut down fetal hemoglobin after birth. ZBTB7A (also called LRF) works alongside BCL11A to suppress fetal hemoglobin. When these factors are reduced, fetal hemoglobin production rises—a discovery that has reshaped modern therapy.
How Does Globin Expression Change Across Developmental Stages?
The body produces different hemoglobins in a carefully timed sequence, matching oxygen needs at each life stage.
Embryonic Globin Expression
In the earliest weeks of development, the yolk sac produces embryonic hemoglobins built from epsilon and zeta chains. These early hemoglobins support the embryo before the liver takes over blood cell production.
Fetal Hemoglobin Dominance and Its Significance
As development continues, gamma-globin production surges and fetal hemoglobin becomes dominant. HbF binds oxygen more tightly than adult hemoglobin, which lets the fetus pull oxygen efficiently across the placenta from the mother’s blood. This higher oxygen affinity is essential for fetal survival and growth.
The Gamma to Beta Globin Switch in Development
Around the time of birth, the gamma to beta globin switch in development begins in earnest. Gamma-globin output declines while beta-globin output climbs, gradually replacing HbF with HbA. By about six months of age, most healthy infants produce mainly adult hemoglobin, with fetal hemoglobin dropping below one percent of the total.
What Mechanisms Drive the Globin Gene Switch?
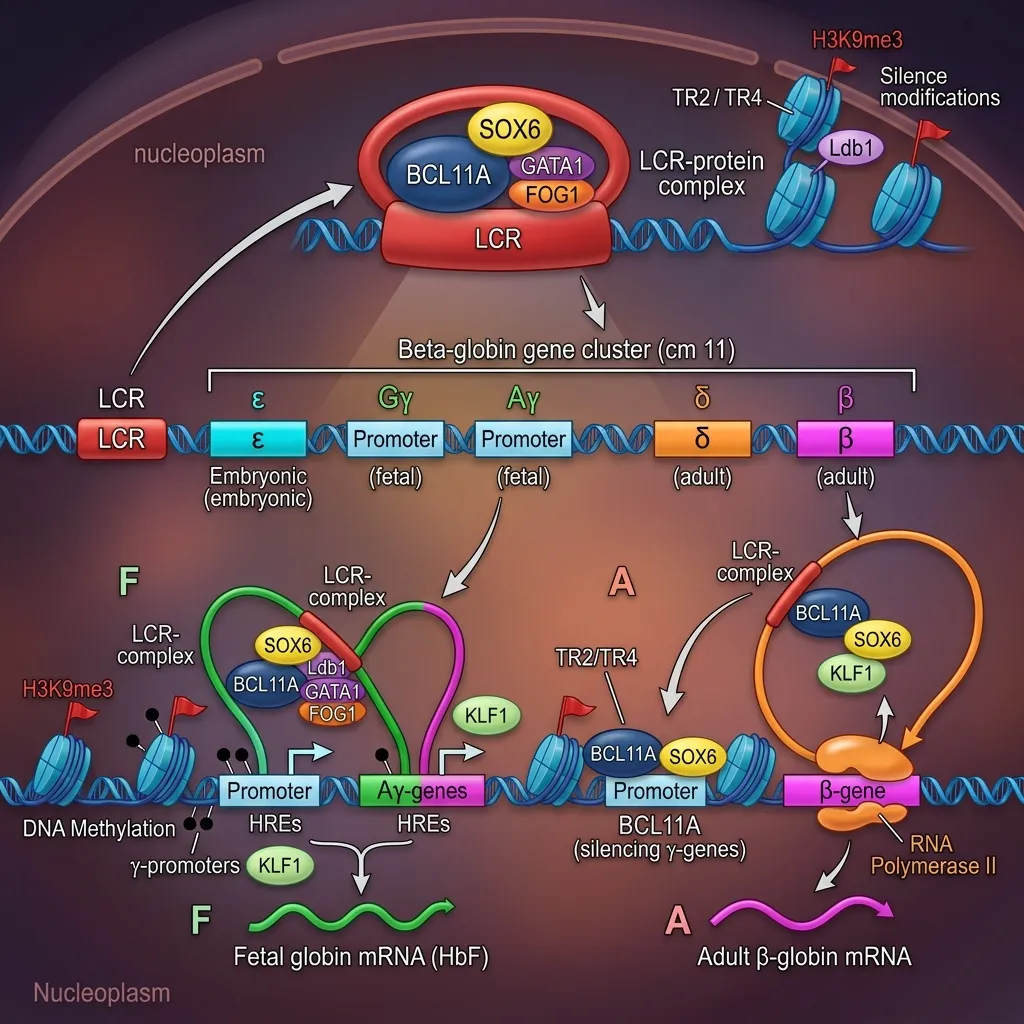 Several layers of control work together to flip the switch from fetal to adult hemoglobin. The hemoglobin gene regulation mechanism explained here combines chromatin changes, transcription factors, and fine-tuning after the gene is read.
Several layers of control work together to flip the switch from fetal to adult hemoglobin. The hemoglobin gene regulation mechanism explained here combines chromatin changes, transcription factors, and fine-tuning after the gene is read.
Chromatin Remodeling and Epigenetic Modifications
Epigenetics refers to chemical changes that alter gene activity without changing the DNA sequence itself. Two key processes shape the globin gene switching mechanism:
- DNA methylation: Adding methyl groups to the gamma-globin gene region helps silence fetal hemoglobin after birth. Removing these groups can reactivate HbF, which is why certain drugs target this process.
- Histone modifications: DNA wraps around proteins called histones. Chemical tags on histones either loosen or tighten this packaging, making genes easier or harder to read. These tags help determine whether gamma or beta genes are active.
Transcriptional Regulation: Key Players and Pathways
At the transcriptional level, the silencers BCL11A and ZBTB7A bind the gamma-globin region and suppress its activity. The LCR, meanwhile, shifts its strongest interaction from the gamma genes to the beta gene as development progresses. This rearrangement of DNA contacts is a central feature of the fetal to adult hemoglobin switching process.
Post-Transcriptional Regulation
Control does not stop once a gene is read. Cells also regulate how stable the resulting messenger RNA is and how efficiently it is translated into protein. These post-transcriptional steps add another layer of precision to the balance between gamma and beta globin output.
Why Is the Globin Gene Switch Clinically Important?
The globin gene switching mechanism is not just a textbook curiosity. It sits at the heart of several inherited blood disorders and shapes how severe those disorders become.
Hereditary Persistence of Fetal Hemoglobin (HPFH)
Hereditary Persistence of Fetal Hemoglobin (HPFH) is a benign condition in which the body keeps producing fetal hemoglobin into adulthood. Normally, HbF switches off after birth, but in HPFH, genetic changes keep it active. Remarkably, HPFH is protective: when it occurs alongside sickle cell disease or beta-thalassemia, the extra fetal hemoglobin softens the severity of those conditions. This natural protection inspired the entire field of HbF-reactivation therapy.
Thalassemia Syndromes and Globin Switching
Thalassemias arise when globin chain production becomes unbalanced. In these conditions, a failure to produce enough of one chain leaves the other in toxic excess. Reactivating fetal hemoglobin can help restore balance because gamma chains can pair with the excess alpha chains seen in beta-thalassemia.
For a deeper look at how unequal chain production drives disease, see this guide on alpha and beta globin chain imbalance. To understand the specific gene defects behind one major form, this detailed resource on beta-thalassemia causes explains the underlying mutations clearly.
Sickle Cell Disease
Sickle cell disease results from a single mutation in the beta-globin gene that produces abnormal hemoglobin S. When oxygen levels drop, these molecules distort red blood cells into rigid sickle shapes that block blood vessels and cause pain. Fetal hemoglobin interferes with this sickling process, so raising HbF levels reduces crises. This is exactly why turning the globin gene switch back on has become a leading treatment strategy.
What Therapies Target the Globin Gene Switch?
Because reactivating fetal hemoglobin eases both sickle cell disease and beta-thalassemia, scientists have developed several strategies to manipulate the globin gene switching mechanism.
Reactivating Fetal Hemoglobin Production
Three drug approaches aim to boost HbF:
- Hydroxyurea: The most established HbF-inducing drug, hydroxyurea increases fetal hemoglobin and reduces the frequency of pain crises in sickle cell patients. It works in part by stimulating gamma-globin expression.
- Butyrate derivatives: These compounds can promote fetal hemoglobin production by influencing histone modifications, loosening the chromatin around gamma genes.
- Decitabine: This drug inhibits DNA methylation, removing the chemical marks that silence gamma-globin and allowing fetal hemoglobin to rise.
Gene Editing Approaches Using CRISPR-Cas9
Gene editing has transformed the field. By using CRISPR-Cas9 to disable BCL11A in a patient’s own blood stem cells, scientists can release the brake on fetal hemoglobin. The edited cells then produce high levels of HbF, which can dramatically reduce symptoms in both sickle cell disease and beta-thalassemia. This approach targets the switch directly rather than relying on daily medication.
Stem Cell Transplantation and Gene Therapy
A bone marrow or stem cell transplant remains the only established cure for severe hemoglobin disorders, replacing defective marrow with healthy donor cells. Gene therapy offers a newer route: doctors collect a patient’s own stem cells, correct or reprogram them in the lab, and return them to the body. Both approaches aim for a lasting solution without lifelong transfusions. For a broader overview of inherited hemoglobin conditions and their care, this guide on hemoglobin synthesis disorders provides helpful context, and this resource on ineffective erythropoiesis explains why these disorders damage red blood cell production.
 The future of treating hemoglobinopathies looks brighter than ever, thanks to ongoing work on the globin gene switching mechanism and its role in hemoglobin production and disease control.
The future of treating hemoglobinopathies looks brighter than ever, thanks to ongoing work on the globin gene switching mechanism and its role in hemoglobin production and disease control.The globin gene switching mechanism stands as one of the most elegant examples of developmental control in human biology. From the early dominance of fetal hemoglobin to the fetal-to-adult hemoglobin switching process, every step after birth is regulated by a precise network of genes, transcription factors, and epigenetic signals. When this system is disrupted, conditions such as beta thalassemia and sickle cell disease can develop.
The encouraging news is that understanding this hemoglobin gene regulation mechanism has opened real therapeutic opportunities. Drugs like hydroxyurea, along with advanced CRISPR-based gene editing, are now used to reactivate fetal hemoglobin (HbF), helping reduce symptoms in patients with hemoglobin disorders. As research continues, gamma to beta globin switching in development may become a key target for fully personalized and potentially curative treatments.
If you or a family member is affected by a hemoglobin disorder, the first step is proper diagnosis through clinical and genetic testing. Consulting a hematology specialist can help you understand treatment options, including emerging therapies that target globin gene regulation. For more information, explore our complete guide to hemoglobin synthesis disorders.
Frequently Asked Questions
1. What is globin gene switching?
Globin gene switching is the developmental process in which the body changes globin gene expression, shifting from embryonic and fetal hemoglobin to adult hemoglobin. This hemoglobin gene regulation mechanism is essential for normal oxygen transport after birth.
2. Why is fetal hemoglobin important?
Fetal hemoglobin (HbF) binds oxygen more efficiently than adult hemoglobin, supporting oxygen transfer from mother to fetus. In disorders like sickle cell disease and beta thalassemia, higher HbF levels improve symptoms and disease outcomes.
3. What causes the gamma-to-beta globin switch in development?
The gamma to beta globin switch in development is controlled by transcription factors such as BCL11A and epigenetic modifications like DNA methylation. These changes silence fetal hemoglobin genes and activate adult hemoglobin production.
4. Can the fetal-to-adult hemoglobin switching process be reversed?
Yes, partially. The fetal to adult hemoglobin switching process can be reactivated using drugs like hydroxyurea or gene-editing therapies that target regulators such as BCL11A, increasing fetal hemoglobin levels in adults.
5. How does hydroxyurea work in reactivating HbF?
Hydroxyurea increases fetal hemoglobin (HbF) production by stimulating gamma-globin expression. This helps reduce red blood cell sickling and improves outcomes in sickle cell disease.
6. What are the risks of therapies targeting globin gene switching?
Therapies targeting globin gene regulation vary in risk. Hydroxyurea may cause bone marrow suppression, while gene therapy and stem cell treatments carry procedural risks requiring specialist monitoring.
7. What is Hereditary Persistence of Fetal Hemoglobin (HPFH)?
HPFH is a genetic condition where fetal hemoglobin continues to be produced into adulthood. It is generally harmless and may reduce severity in conditions like sickle cell disease and beta thalassemia.
8. How does globin gene switching relate to thalassemia?
In thalassemia, an imbalance in globin chain production causes anemia. Reactivating fetal hemoglobin through manipulation of the globin gene switching mechanism helps restore balance and reduce symptoms.
9. What is the role of epigenetics in hemoglobin gene regulation?
Epigenetics controls gene activity without changing the DNA sequence. In hemoglobin regulation, epigenetic marks silence fetal hemoglobin genes after birth and are key targets for therapies aiming to reverse this process.
10. Where can I find more information about hemoglobin disorders?
You can explore detailed resources on hemoglobin disorders, including causes, diagnosis, and treatment strategies, in comprehensive guides focused on hemoglobin synthesis disorders.