


Learn why beta thalassemia anemia develops, how HBB gene mutations disrupt hemoglobin production, and why red blood cells are destroyed prematurely. This guide explains the causes, symptoms, diagnosis, treatment options, and practical strategies for managing the condition effectively.
Beta thalassemia causes anemia because mutations in the HBB gene reduce beta globin production. Excess alpha chains build up, damage developing red blood cells, and destroy them early through ineffective erythropoiesis.
Beta thalassemia anemia is one of the most common inherited blood disorders in the world, yet many people still struggle to understand why it happens. The short version is simple: the body cannot make enough healthy hemoglobin, so red blood cells fall short on their main job—carrying oxygen.
But the full story runs deeper than that. Anemia in beta thalassemia is not caused by a single broken step. It comes from a chain of events that starts in the genes, moves through the bone marrow, and ends with red blood cells dying before they can mature. Understanding this process helps patients, caregivers, and students make sense of symptoms, test results, and treatment choices.
The severity of anemia varies depending on the type of beta thalassemia and the specific genetic mutation involved. Some individuals experience only mild symptoms, while others require lifelong blood transfusions and specialized medical care. Without effective treatment, chronic anemia can affect growth, organ function, energy levels, and overall quality of life. Early diagnosis through blood tests and genetic screening allows healthcare providers to begin appropriate management before serious complications develop. Advances in treatments, including iron chelation therapy, stem cell transplantation, and gene therapy, are also improving long-term outcomes for many patients. Learning how beta thalassemia causes anemia empowers individuals to recognize symptoms early, understand available therapies, and participate more actively in their care.
In this guide, you’ll learn exactly why beta thalassemia causes anemia, how beta thalassemia causes anemia at the cellular level, what symptoms to watch for, and which treatments can help. By the end, you’ll have a clear, complete picture of this condition and the steps that can improve quality of life.
What Is Beta Thalassemia Anemia?
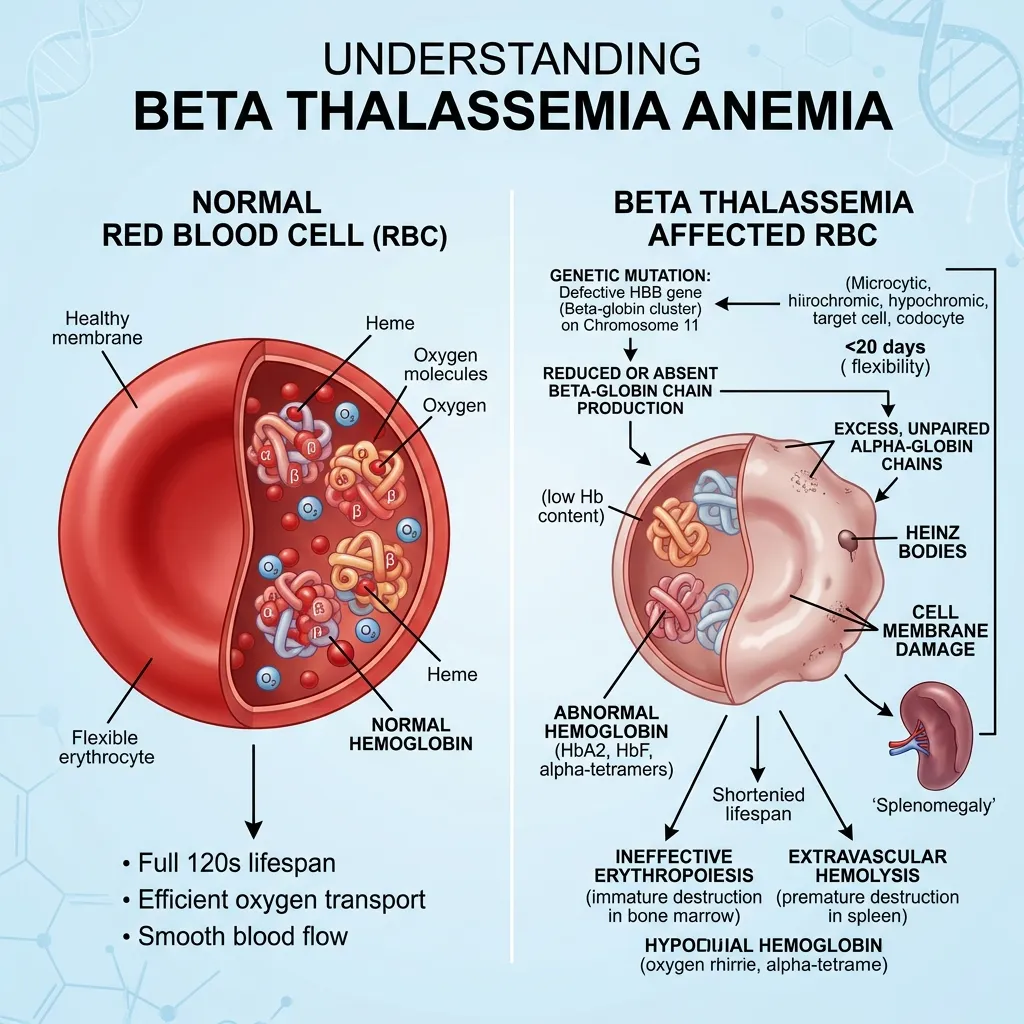
Beta thalassemia anemia is a type of inherited anemia that develops when the body cannot produce enough beta globin chains. These chains are a key building block of hemoglobin, the protein inside red blood cells that carries oxygen throughout the body.
Normal adult hemoglobin (HbA) is made of two alpha globin chains and two beta globin chains. These chains must be produced in nearly equal amounts. When beta chain production drops, the balance breaks, and red blood cells suffer the consequences. You can read more about how these chains work together in this guide on the alpha vs beta globin function difference.
The condition comes in several forms, depending on how severely beta globin production is affected:
- Beta thalassemia minor (trait): Mild, often symptomless. Many carriers never know they have it.
- Beta thalassemia intermedia: Moderate anemia that may require occasional treatment.
- Beta thalassemia major: Severe, transfusion-dependent anemia that appears in early childhood.
The severity of beta thalassemia anemia depends largely on how much functional beta globin the body can still produce.
Why Does Beta Thalassemia Cause Anemia?
To understand why beta thalassemia causes anemia, you need to start with genetics. Beta thalassemia results from mutations in the HBB gene on chromosome 11. This gene carries the instructions for making beta globin chains.
When the HBB gene is mutated, beta globin production drops or stops entirely. Because hemoglobin needs two beta chains to form correctly, less beta globin means less healthy hemoglobin. With fewer functional hemoglobin molecules, red blood cells cannot carry enough oxygen, and anemia develops.
There’s a second, more damaging problem at play. The body keeps making alpha chains at a normal rate, even when beta chains run short. This creates an imbalance, leaving excess alpha chains with no beta partners to pair with. These unpaired alpha chains are highly unstable and toxic to developing red blood cells. You can explore this mechanism in detail in this guide on alpha and beta globin chain imbalance.
So the answer to “why does beta thalassemia cause anemia” has two parts:
- Reduced hemoglobin production: Fewer beta chains mean less functional hemoglobin.
- Toxic alpha chain buildup: Excess alpha chains damage and destroy red blood cells before they mature.
Together, these two problems create the chronic, often severe anemia seen in beta thalassemia.
How Beta Thalassemia Causes Anemia at the Cellular Level
Now let’s look closer at how beta thalassemia causes anemia inside the bone marrow. This is where the real damage happens.
The Role of Excess Alpha Chains
When beta chains are in short supply, free alpha chains pile up inside developing red blood cells. These chains cannot form stable hemoglobin on their own. Instead, they clump together into toxic aggregates.
These aggregates cause serious harm. They damage cell membranes, trigger oxidative stress, and disrupt the normal process of red blood cell maturation. As a result, many red blood cell precursors die inside the bone marrow before they ever reach the bloodstream.
Ineffective Erythropoiesis
This premature destruction of red blood cells in the bone marrow is called ineffective erythropoiesis. It’s one of the central mechanisms behind anemia in beta thalassemia.
Here’s the vicious cycle it creates:
- The bone marrow senses low oxygen and tries to make more red blood cells.
- Most of these new cells are damaged by excess alpha chains.
- The damaged cells die before maturing.
- Anemia persists, so the bone marrow works even harder.
The marrow becomes hyperactive, yet very few healthy red blood cells reach circulation. To understand this process in depth, see this detailed guide on ineffective erythropoiesis.
Shortened Red Blood Cell Survival
The few red blood cells that do make it into the bloodstream don’t last long. Damaged and unstable, they are removed early by the spleen. Healthy red blood cells normally survive about 120 days, but in beta thalassemia, their lifespan is much shorter. This adds another layer to the anemia.
What Are the Symptoms of Beta Thalassemia Anemia?
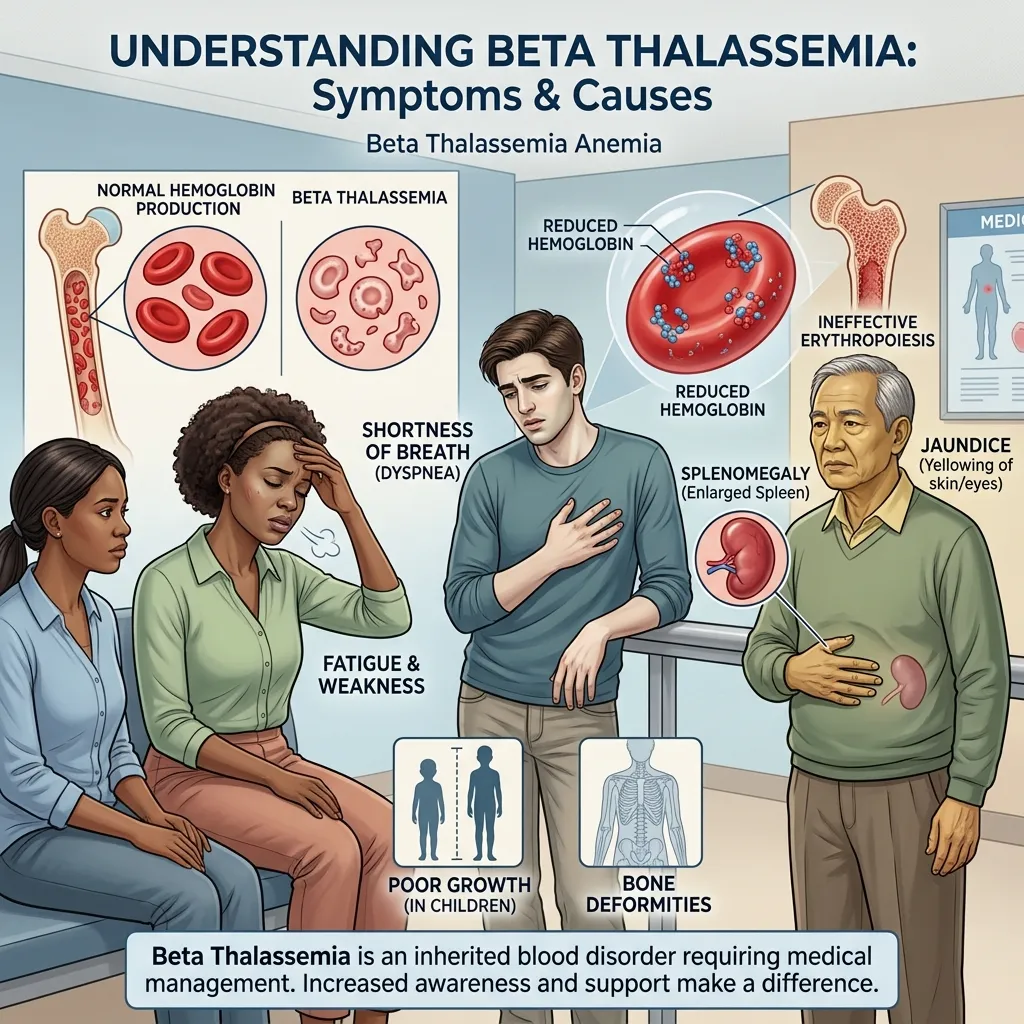 The symptoms of beta thalassemia anemia stem directly from low oxygen delivery and the body’s struggle to compensate. Severity varies widely depending on the form of the disease.
The symptoms of beta thalassemia anemia stem directly from low oxygen delivery and the body’s struggle to compensate. Severity varies widely depending on the form of the disease.
Common symptoms include:
- Persistent fatigue and weakness
- Pale or yellowish skin
- Shortness of breath
- Dizziness and headaches
- Poor exercise tolerance
- Rapid heartbeat
In more severe cases, patients may experience:
- Enlarged spleen (splenomegaly)
- Enlarged liver (hepatomegaly)
- Bone deformities, especially in the face and skull
- Delayed growth and puberty in children
- Iron overload, even without frequent transfusions
These severe symptoms reflect the long-term strain of chronic anemia and ineffective erythropoiesis on the body.
How Is Anemia in Beta Thalassemia Diagnosed?
Doctors use several tests to confirm beta thalassemia anemia and measure its severity. According to the Centers for Disease Control and Prevention, early diagnosis is key to managing inherited blood disorders effectively.
- Complete blood count (CBC): Often reveals small, pale red blood cells (microcytic, hypochromic anemia) and low hemoglobin levels.
- Peripheral blood smear: Shows abnormally shaped red blood cells, including target cells.
- Hemoglobin electrophoresis: Separates hemoglobin types and helps distinguish beta thalassemia from other disorders. It often shows elevated HbA2 and HbF.
- Genetic testing: Confirms specific HBB gene mutations and carrier status.
These tests together build a complete picture of the condition and guide treatment decisions.
How Is Beta Thalassemia Anemia Treated?
While beta thalassemia anemia cannot always be cured, several treatments can manage symptoms and improve quality of life. The right approach depends on disease severity.
Blood Transfusions
Regular blood transfusions are the cornerstone of treatment for severe beta thalassemia. They provide healthy red blood cells, raise hemoglobin levels, and suppress the harmful overactivity of the bone marrow.
Iron Chelation Therapy
Frequent transfusions and increased iron absorption can lead to dangerous iron overload. Iron chelation therapy uses medication to remove excess iron and protect organs like the heart and liver.
Folic Acid Supplementation
Folic acid supports red blood cell production and is often recommended to help the bone marrow function as effectively as possible.
Bone Marrow or Stem Cell Transplant
A stem cell transplant is currently the only established cure for severe beta thalassemia. It replaces defective bone marrow with healthy donor cells, though it carries significant risks and requires a suitable donor.
Emerging Gene Therapies
Newer treatments aim to correct the underlying genetic defect or reactivate fetal hemoglobin (HbF) production. Gene-editing approaches, including CRISPR-based therapies, have shown promising results in reducing transfusion dependence for some patients.

Living with Beta Thalassemia Anemia
 Living with beta thalassemia anemia requires more than treating low hemoglobin levels. It involves a long-term approach that combines medical care, healthy lifestyle habits, and emotional support. While the condition is lifelong, many people can lead active, productive lives with proper management.
Living with beta thalassemia anemia requires more than treating low hemoglobin levels. It involves a long-term approach that combines medical care, healthy lifestyle habits, and emotional support. While the condition is lifelong, many people can lead active, productive lives with proper management.
Conclusion
Beta thalassemia anemia develops from a clear chain of events. Mutations in the HBB gene reduce beta globin production, which lowers healthy hemoglobin levels. At the same time, excess alpha chains accumulate, damage red blood cells, and trigger ineffective erythropoiesis. The result is chronic anemia that ranges from mild to severe.
Understanding why beta thalassemia causes anemia—and how beta thalassemia causes anemia at the cellular level—empowers patients and families to make informed decisions about diagnosis, treatment, and daily care.
If you or a loved one may be affected by a hemoglobin disorder, the best first step is proper diagnosis through clinical and genetic testing. Speak with a hematology specialist about your options, and keep learning through trusted resources on inherited blood conditions.
Frequently Asked Questions
1. What is beta thalassemia anemia?
Beta thalassemia anemia is an inherited blood disorder where mutations in the HBB gene reduce beta globin production. This lowers healthy hemoglobin levels and causes red blood cells to be destroyed early, leading to chronic anemia.
2. Why does beta thalassemia cause anemia?
Beta thalassemia causes anemia for two reasons. First, reduced beta globin means less functional hemoglobin. Second, excess alpha chains build up, damage red blood cells, and destroy them before they mature in the bone marrow.
3. How does beta thalassemia cause anemia at the cellular level?
Anemia in beta thalassemia develops when unpaired alpha chains clump together inside developing red blood cells. These toxic aggregates damage the cells and cause them to die early through a process called ineffective erythropoiesis.
4. Is beta thalassemia anemia genetic?
Yes. Beta thalassemia is an inherited autosomal recessive disorder. A child must inherit a mutated HBB gene from both parents to develop a severe form of the disease.
5. What are the main symptoms of anemia in beta thalassemia?
Common symptoms include fatigue, pale skin, shortness of breath, dizziness, and a rapid heartbeat. Severe cases can cause an enlarged spleen, bone deformities, and delayed growth in children.
6. How is beta thalassemia anemia diagnosed?
Doctors use a complete blood count, peripheral blood smear, hemoglobin electrophoresis, and genetic testing. These tests confirm the diagnosis, measure severity, and identify the specific gene mutation involved.
7. Can beta thalassemia anemia be cured?
A bone marrow or stem cell transplant is currently the only established cure for severe cases. Emerging gene therapies, including CRISPR-based treatments, also show promise in reducing transfusion dependence.
8. What is the difference between beta thalassemia minor and major?
Beta thalassemia minor (trait) is mild and often symptomless. Beta thalassemia major is severe, appears in early childhood, and requires lifelong blood transfusions and ongoing medical care.
9. Why does beta thalassemia cause iron overload?
The body increases iron absorption to compensate for anemia, and frequent transfusions add more iron. Because red blood cell production remains defective, this excess iron builds up and can damage organs over time.
10. How is beta thalassemia anemia different from iron deficiency anemia?
Both cause small, pale red blood cells, but the causes differ. Iron deficiency anemia results from low iron, while beta thalassemia anemia is genetic. Taking iron supplements does not treat beta thalassemia and may cause harmful iron overload.