


Thalassemia genetic causes are linked to inherited mutations in hemoglobin genes passed from parents to children. These genetic changes reduce normal hemoglobin production, leading to anemia and related health issues, with severity depending on the type and combination of inherited genes.
The Genetic Basis of Thalassemia
To understand how thalassemia develops, we need to look at its root cause inside human DNA. At the center of this condition is hemoglobin, the protein responsible for carrying oxygen in red blood cells. The underlying issue comes from changes in the genes that control hemoglobin production, known as thalassemia genetic causes.
Hemoglobin is made of two types of protein chains—alpha and beta. A precise balance between these chains is essential for normal red blood cell function. When genetic mutations affect either chain, the body cannot produce healthy hemoglobin, leading to early breakdown of red blood cells and anemia.
These genetic instructions are stored in genes within chromosomes, which we inherit from both parents. A mutation is simply a change in this genetic code. In thalassemia, such mutations disrupt the production of alpha or beta globin chains, and the severity of the disease depends on how many genes are affected.
Types of Genetic Effects
| Mutation Type | Effect |
|---|---|
| Alpha gene mutation | Reduced alpha chain production |
| Beta gene mutation | Reduced or absent beta chain production |
| Combined mutations | Severe imbalance in hemoglobin |
Types of Thalassemia and Their Genetic Roots
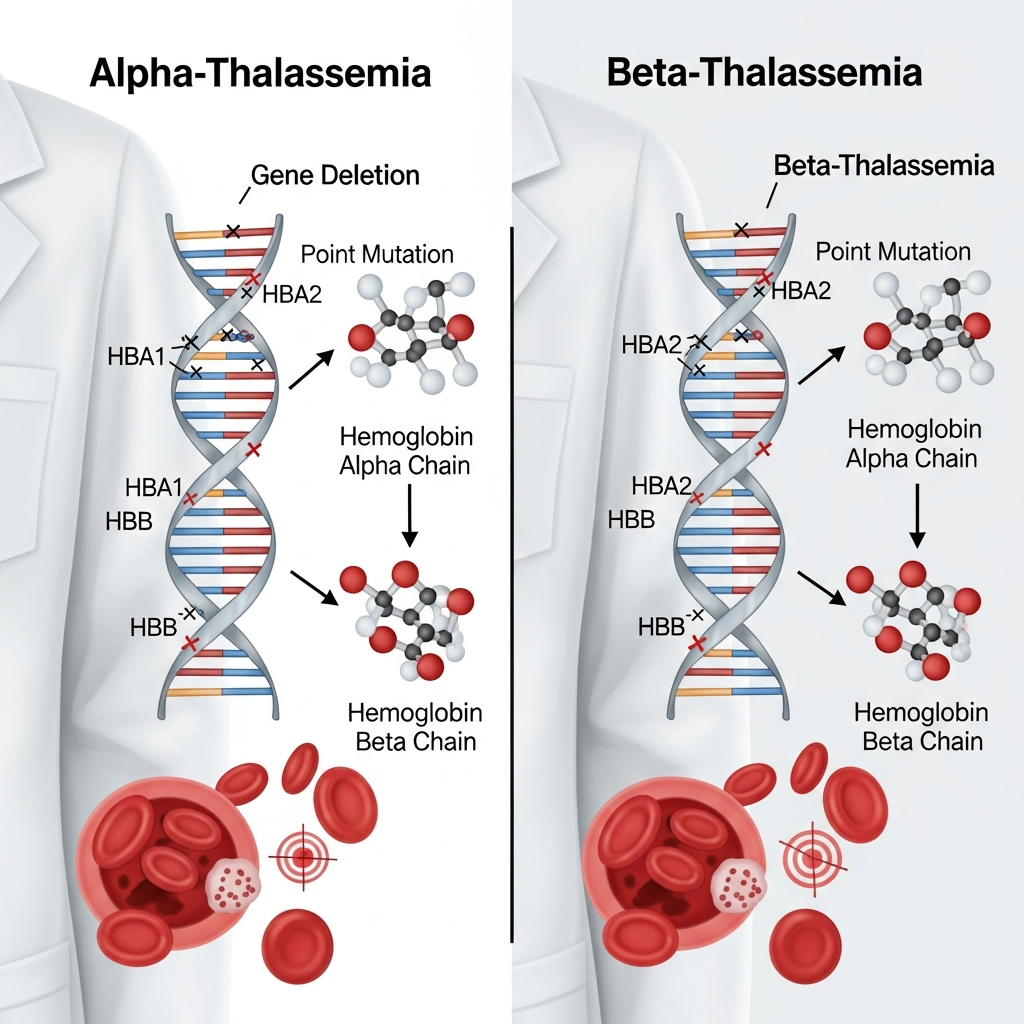 Because hemoglobin depends on two different protein chains—alpha and beta—thalassemia is broadly classified into two main types. Each type is linked to specific genetic changes on different chromosomes. Understanding these variations helps explain the severity and symptoms seen in patients and is a key part of understanding thalassemia genetic causes.
Because hemoglobin depends on two different protein chains—alpha and beta—thalassemia is broadly classified into two main types. Each type is linked to specific genetic changes on different chromosomes. Understanding these variations helps explain the severity and symptoms seen in patients and is a key part of understanding thalassemia genetic causes.
Alpha-Thalassemia: Genetic Defects on Chromosome 16
Alpha-thalassemia occurs when there is a disruption in the production of alpha-globin chains. The genes responsible for these chains are located on chromosome 16.
Gene Deletion and Severity
Each person inherits four alpha-globin genes—two from each parent. The condition’s severity depends on how many of these genes are missing or damaged:
- 1 gene affected: Silent carrier (no symptoms)
- 2 genes affected: Alpha-thalassemia trait (mild anemia)
- 3 genes affected: Hemoglobin H disease (moderate to severe anemia)
- 4 genes affected: Alpha-thalassemia major (hydrops fetalis, usually fatal before or shortly after birth)
This stepwise pattern clearly shows how thalassemia genetic causes directly influence disease severity.
Clinical Impact
In milder cases, individuals may live normal lives with minimal symptoms. However, in more severe forms like HbH disease, patients may experience:
- Chronic fatigue and weakness
- Enlarged spleen
- Bone abnormalities
- Need for occasional blood transfusions
The most severe form prevents the fetus from producing functional hemoglobin, making survival extremely difficult.
Beta-Thalassemia: Genetic Defects on Chromosome 11
Beta-thalassemia results from mutations affecting beta-globin chain production. The responsible genes are located on chromosome 11.
Point Mutations and Their Effect
Unlike alpha-thalassemia, which often involves gene deletions, beta-thalassemia is usually caused by point mutations—small errors in the DNA sequence that disrupt gene function.
Each person inherits two beta-globin genes—one from each parent. The condition depends on whether:
- One gene is affected → mild condition
- Both genes are affected → severe condition
These mutations are a major example of thalassemia genetic causes at the molecular level.
Clinical Manifestations
- Beta-thalassemia minor (trait): Usually mild or no symptoms, slight anemia may occur
- Beta-thalassemia intermedia: Moderate symptoms, occasional treatment needed
- Beta-thalassemia major (Cooley’s anemia): Severe form requiring lifelong blood transfusions and medical care
Without treatment, the major form can lead to serious complications such as growth failure, bone deformities, and organ damage due to iron overload.
Key Genetic Differences Between Alpha and Beta Types
| Feature | Alpha-Thalassemia | Beta-Thalassemia |
|---|---|---|
| Chromosome | 16 | 11 |
| Type of Mutation | Gene deletions | Point mutations |
| Number of Genes Involved | 4 genes | 2 genes |
| Severity Pattern | Based on number of missing genes | Based on mutation type and gene function |
Why These Genetic Differences Matter
 Understanding these variations in thalassemia genetic causes is essential for:
Understanding these variations in thalassemia genetic causes is essential for:
- Early and accurate diagnosis
- Genetic counseling for families
- Predicting disease severity
- Planning appropriate long-term treatment

Common Signs in Infants and Children
Children born with severe forms of the disorder usually appear healthy at birth. However, within the first two years of life, the effects of inadequate hemoglobin production begin to show.
Fatigue and Weakness
Because the body’s tissues are starving for oxygen, children often experience extreme tiredness. They may lack the energy to play and can tire quickly during normal activities.
Pale Skin and Jaundice
The shortage of red blood cells leads to a noticeable paleness. Additionally, as the body attempts to break down defective red blood cells, a substance called bilirubin builds up. This causes jaundice, a yellowish tint to the skin and the whites of the eyes.
Poor Feeding and Growth Delays
Oxygen is required for healthy cellular growth and metabolism. Infants with severe forms of the disorder often struggle to feed properly, leading to slow weight gain and delayed physical development.
Enlarged Spleen or Liver
The spleen acts as a filter for the blood. Because the red blood cells are defective, the spleen works overtime to clear them out. This heavy workload causes the spleen—and sometimes the liver—to enlarge significantly, which can be felt by a doctor during a physical exam.
Symptoms in Adults with Milder Forms
People with the minor trait often do not show symptoms during childhood. As adults, they might experience mild fatigue or feel slightly dizzy during intense physical exertion. These mild symptoms are frequently discovered incidentally during routine blood work for other conditions.
The Importance of Early Detection and Diagnosis
Detecting the disorder early allows doctors to intervene before severe complications, such as bone deformities or heart failure, can occur. If you notice any of these symptoms in your child, consult a healthcare provider promptly. For a comprehensive overview of clinical signs, you can review this reputable medical source on thalassemia symptoms.
Diagnostic Approaches: Confirming Thalassemia
If a doctor suspects a patient has the disorder based on physical symptoms or family history, several specific tests are used to confirm the diagnosis.
Blood Tests
The first step is typically a Complete Blood Count (CBC). This test measures the amount of hemoglobin and the different types of blood cells. In patients with the disorder, red blood cells often appear smaller and paler than normal under a microscope.
If the CBC indicates abnormalities, a doctor will order a hemoglobin electrophoresis test. This specialized test separates the different types of hemoglobin in the blood, allowing doctors to see exactly which proteins are missing or present in abnormal amounts.
Genetic Testing for Definitive Diagnosis
While blood tests can indicate the presence of the disorder, genetic testing provides the most definitive answer. By analyzing a sample of DNA—usually obtained through a simple blood draw—laboratories can pinpoint the exact mutations on chromosome 11 or 16. This confirms the specific type and severity of the condition.
Prenatal Diagnosis Options
For expectant parents who know they are carriers, prenatal testing is available. Procedures such as Chorionic Villus Sampling (CVS) or amniocentesis can extract DNA from the placenta or amniotic fluid. This allows doctors to test the fetus for genetic mutations well before birth, giving parents time to prepare and consult with medical specialists.
Living with Thalassemia: Management and Treatment
 While the severe forms of this genetic disorder pose significant challenges, modern medicine offers highly effective ways to manage the condition.
While the severe forms of this genetic disorder pose significant challenges, modern medicine offers highly effective ways to manage the condition.
Blood Transfusions
For individuals with major forms of the disorder, regular blood transfusions are the cornerstone of treatment. These transfusions, often required every few weeks, provide the body with a fresh supply of healthy red blood cells, instantly alleviating the symptoms of anemia.
Chelation Therapy
While blood transfusions are life-saving, they introduce a secondary problem: iron overload. The human body has no natural way to eliminate excess iron from transfused blood. Over time, iron builds up and can cause fatal damage to the heart and liver. Chelation therapy involves taking specific medications that bind to the excess iron, allowing the body to excrete it through urine or stool.
Bone Marrow Transplantation
Currently, the only potential cure for the disorder is a bone marrow transplant (also known as a stem cell transplant). This procedure replaces the patient’s defective bone marrow with healthy stem cells from a compatible donor, usually a sibling. If successful, the new bone marrow will begin producing normal, healthy red blood cells. However, this is a high-risk procedure and is not suitable for everyone.
Lifestyle Adjustments
Patients are encouraged to adopt specific lifestyle changes. A balanced diet, avoiding iron supplements (unless specifically instructed by a hematologist), and taking folic acid to help build red blood cells are all standard recommendations. Regular check-ups are also mandatory to monitor organ health.
Prevention and Genetic Counseling
Because this is an inherited condition, prevention relies heavily on education and proactive medical screening.
Pre-Marital and Pre-Conception Screening
In many parts of the world where the disorder is highly prevalent, pre-marital screening programs are standard practice. A simple blood test can reveal if prospective partners are both carriers. Couples can then seek counseling before making decisions about having children.
The Role of Genetic Counselors
Genetic counselors are specially trained professionals who help individuals navigate the complex world of inherited diseases. They explain the risks, interpret test results, and discuss all available reproductive options, such as in-vitro fertilization (IVF) with preimplantation genetic testing to ensure embryos are free of the mutation.
Reducing the Incidence of Severe Forms
Through widespread awareness and accessible screening programs, several countries have successfully reduced the number of children born with severe forms of the disease. Education remains the most powerful tool in managing the global impact of this condition.
Navigating the Future with Knowledge
Understanding the biological roots of this disorder changes how we view it. It is not an unpredictable illness, but rather a specific genetic error involving chromosomes and hemoglobin. Grasping the exact thalassemia genetic mutation causes removes the stigma and empowers families to take control of their health through screening and early intervention.
Medical science continues to advance rapidly. Gene therapy, a process that aims to correct the defective DNA within the patient’s own body, is currently undergoing clinical trials and showing immense promise. As research progresses, the goal of a universal, accessible cure moves closer to reality. Until then, awareness, proper medical management, and genetic counseling remain the strongest defenses against this enduring genetic condition.