


What exactly is Beta Thalassemia and what are its types?
Beta Thalassemia is a genetic blood disorder that limits the body’s ability to produce hemoglobin. Hemoglobin is the iron-rich protein in red blood cells that carries oxygen to tissues. Medical professionals classify Beta Thalassemia into three main types based on severity: major, intermedia, and minor. Beta Thalassemia major (Cooley’s anemia) requires lifelong blood transfusions. Beta Thalassemia intermedia causes moderate anemia, while Beta Thalassemia minor (trait) often presents with minimal symptoms. You can learn more about carrier status in this beta thalassemia trait carrier guide. According to the World Health Organization (WHO, 2022), these inherited blood disorders are most prevalent in Mediterranean, Middle Eastern, and Southeast Asian populations.
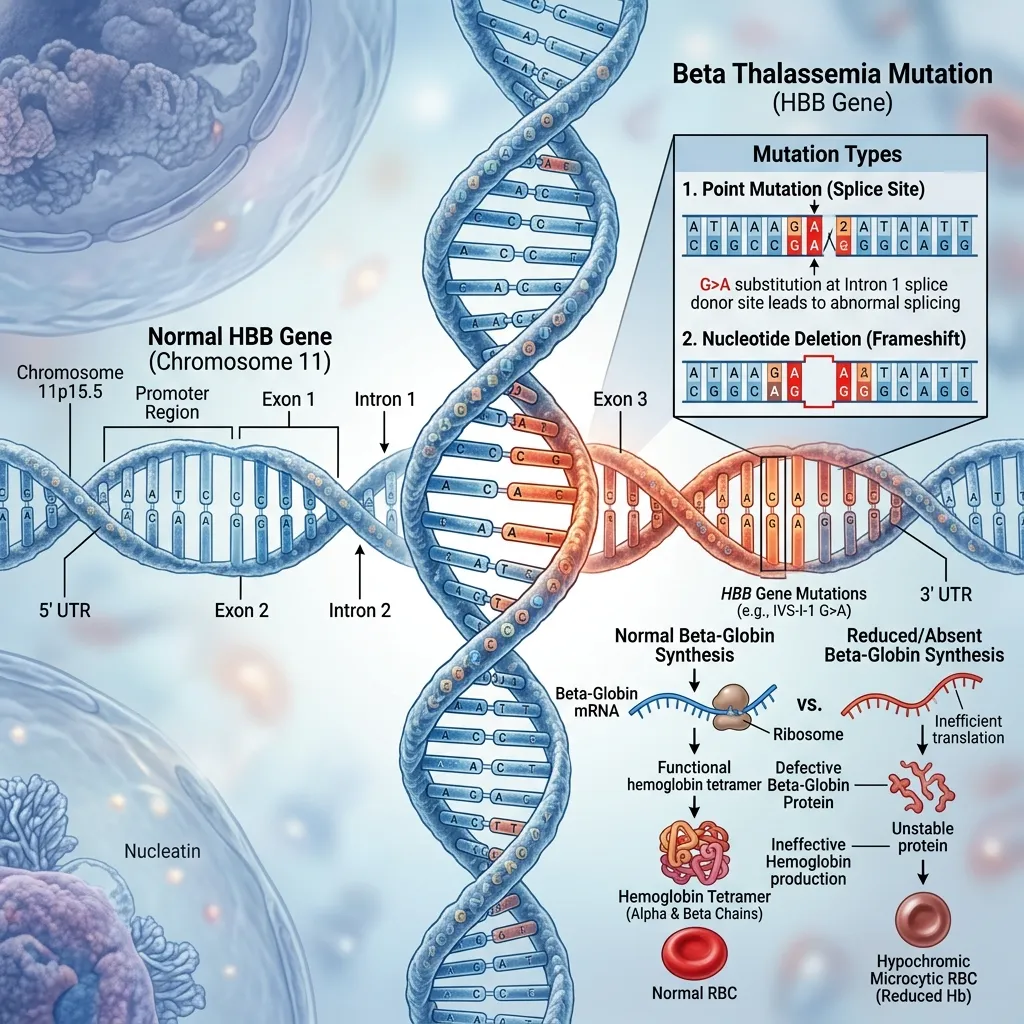
How does a genetic mutation in beta thalassemia affect red blood cells?
A specific genetic mutation in beta thalassemia targets the HBB gene on chromosome 11. The HBB gene provides the instructions for making beta-globin. When this gene mutates, the body produces fewer or no beta-globin chains. Without sufficient beta-globin, red blood cells become small, pale, and highly fragile. These defective cells die prematurely, leading to severe chronic anemia. The bone marrow attempts to compensate by producing more red blood cells, but the underlying genetic defect renders these new cells equally ineffective.
How are inherited blood disorders passed down through families?
Beta Thalassemia follows an autosomal recessive inheritance pattern. This means a child must inherit two mutated HBB genes—one from each parent—to develop the severe form of the disease. If a person inherits only one mutated gene, they become a carrier. Carriers typically live normal lives but can pass the mutated gene to their offspring. Genetic counseling is vital for prospective parents with a family history of inherited blood disorders, as it helps them understand transmission risks and explore family planning options.
What are the long-term consequences of hemoglobin gene defects?
The long-term consequences of hemoglobin gene defects extend far beyond basic anemia. The body’s constant struggle to produce healthy red blood cells leads to ineffective erythropoiesis. This chronic state forces the bone marrow to expand, which can cause severe bone deformities. Additionally, patients absorb excess iron from their diet, and frequent blood transfusions compound this iron overload. Excess iron accumulates in the heart, liver, and endocrine system, causing fatal organ damage if left untreated.
What role do platelets play in patients with Beta Thalassemia?
What are platelets and what is their normal function?
Platelets, or thrombocytes, are specialized cell fragments produced in the bone marrow. Their primary function is hemostasis, which is the process of stopping bleeding at the site of a blood vessel injury. When a blood vessel tears, platelets rush to the site, stick together, and form a plug. A normal platelet count ranges from 150,000 to 450,000 platelets per microliter of blood. To understand how platelets interact with other blood components, you can review this comprehensive different kinds of blood diseases guide.
Why do Beta Thalassemia patients develop thrombocytopenia?
Thrombocytopenia refers to a dangerously low platelet count. In patients with Beta Thalassemia, several factors cause platelet levels to plummet:
- Hypersplenism: The spleen normally filters old blood cells. In Beta Thalassemia, the spleen works overtime and becomes massively enlarged. This enlarged spleen traps and destroys healthy platelets.
- Bone marrow suppression: The bone marrow prioritizes red blood cell production to combat anemia, often at the expense of platelet production. Furthermore, iron overload can physically damage the bone marrow space.
- Liver disease: Iron toxicity frequently damages the liver. A damaged liver produces less thrombopoietin, the hormone responsible for stimulating platelet production.
- Medications: Certain drugs used to treat complications can suppress bone marrow function. It is important to know the thalassemia minor drugs to avoid to prevent accidental marrow suppression.
What are the clinical manifestations of a low platelet count?
When a patient’s platelet count drops below safe thresholds, their bleeding risk increases dramatically. Clinical signs of thrombocytopenia include petechiae (tiny red or purple spots on the skin), purpura (larger bruises), epistaxis (frequent nosebleeds), and menorrhagia (abnormally heavy menstrual periods). Early detection is paramount. If a patient with Beta Thalassemia begins exhibiting unexplained bruising or prolonged bleeding, their medical team must intervene immediately to prevent internal hemorrhaging.
Why are platelet transfusions critical for Beta Thalassemia management?

What is a platelet transfusion and what types are available?
A platelet transfusion is a medical procedure that delivers concentrated donor platelets directly into a patient’s bloodstream. The primary goal is to prevent or treat severe bleeding episodes caused by low platelet counts. Blood centers prepare platelet products in two main ways. Random donor platelets are pooled from multiple whole blood donations. Apheresis platelets are collected from a single donor using a specialized machine that extracts only the platelets and returns the remaining blood components to the donor. According to the American Red Cross, apheresis platelets reduce the patient’s exposure to multiple donors, thereby lowering the risk of immune reactions.
What are the primary indications for a platelet transfusion?
Doctors do not prescribe platelet transfusions lightly. They reserve this intervention for specific clinical scenarios:
- Active bleeding: If a patient with significant thrombocytopenia experiences active, uncontrollable bleeding, a platelet transfusion is a life-saving necessity.
- Prophylaxis before invasive procedures: Surgeons require patients to have a minimum platelet count before operating. Transfusions are administered prior to major surgeries or organ biopsies to prevent surgical hemorrhage.
- Severe symptomatic thrombocytopenia: Even without active bleeding, exceptionally low platelet counts (often below 10,000 per microliter) warrant prophylactic transfusions to prevent spontaneous internal bleeding, particularly in the brain.
How is the platelet transfusion process administered?
The transfusion process requires strict protocols to ensure patient safety. First, the laboratory performs pre-transfusion testing to match the donor platelets with the patient’s ABO blood type and Rh factor. Once matched, healthcare professionals administer the platelets intravenously over a period of 15 to 30 minutes. Medical staff monitor the patient closely for adverse reactions. Doctors expect a measurable platelet increment (a rise in the platelet count) within an hour of the transfusion. The lifespan of transfused platelets is short, typically lasting only a few days in the patient’s bloodstream.
What risks and complications are associated with platelet transfusions?
While lifesaving, platelet transfusions carry specific medical risks. The most common complications are allergic reactions, ranging from mild hives to severe anaphylaxis. Patients may also experience febrile non-hemolytic transfusion reactions, characterized by sudden fevers and chills. Rare but severe risks include Transfusion-Related Acute Lung Injury (TRALI) and Transfusion-Associated Circulatory Overload (TACO). Repeated transfusions can lead to alloimmunization, where the patient’s immune system creates antibodies against donor platelets, making future transfusions ineffective. Finally, while modern screening is highly rigorous, a minute risk of transfusion-transmitted infections remains (According to the National Institutes of Health).
What are the management strategies for Beta Thalassemia beyond transfusions?

Why do patients need regular red blood cell transfusions?
While platelet transfusions address clotting problems and help manage bleeding risks, regular red blood cell transfusions remain the cornerstone of treatment for Beta Thalassemia major. These transfusions are essential because they provide healthy red blood cells containing functional hemoglobin, which improves oxygen delivery throughout the body. By maintaining stable hemoglobin levels, transfusions help suppress the patient’s ineffective bone marrow activity, prevent severe anemia-related complications, and support normal physical growth and development, especially in children.
However, long-term transfusion therapy comes with a significant challenge—iron overload. Each red blood cell transfusion introduces excess iron into the body, and since the human body has no natural way to eliminate it, iron gradually accumulates in vital organs such as the heart, liver, and endocrine glands. This is where iron chelation therapy becomes essential in comprehensive Beta Thalassemia management strategies. Chelation medications bind to excess iron and help remove it through urine or feces, preventing life-threatening complications such as heart failure, liver cirrhosis, and hormonal dysfunction.
Effective disease management in Beta Thalassemia requires a balance between transfusion support and iron control, making long-term monitoring a critical part of patient care. Regular blood tests, MRI-based iron assessments, and dose adjustments are necessary to ensure treatment safety and effectiveness.
When do doctors recommend a splenectomy for Beta Thalassemia?
In some patients, the spleen becomes abnormally enlarged due to excessive destruction of blood cells, a condition known as hypersplenism. When this occurs, the spleen begins to break down not only damaged cells but also healthy red blood cells and platelets at an accelerated rate. This leads to worsening anemia, increased transfusion needs, and persistently low platelet counts, despite regular treatment.
In such cases, doctors may recommend a splenectomy, which is the surgical removal of the spleen. This procedure can help stabilize blood counts, reduce the frequency of red blood cell transfusions, and improve platelet levels in certain patients. However, splenectomy is not a first-line treatment and is usually considered only when hypersplenism severely affects quality of life or treatment response.
Because the spleen plays a key role in immune defense, especially against encapsulated bacteria, patients who undergo splenectomy face a higher risk of serious infections. To reduce this risk, they must receive specific vaccinations before surgery and may require long-term prophylactic antibiotics. Regular medical follow-up is also essential to ensure early detection and prevention of infections.
Conclusion
Managing Beta Thalassemia requires a comprehensive and long-term treatment approach that goes beyond a single therapy. Regular red blood cell transfusions remain essential for maintaining healthy hemoglobin levels and preventing severe anemia, while iron chelation therapy helps control dangerous iron overload. In certain cases, additional interventions such as splenectomy may be needed to manage complications like hypersplenism and improve blood cell counts.
Overall, effective Beta Thalassemia management strategies focus on balancing transfusion support, preventing complications, and closely monitoring the patient’s condition. With proper medical care, timely interventions, and ongoing follow-up, patients can achieve better quality of life and reduced risk of long-term complications.
Frequently Asked Questions
1. What are the main beta thalassemia causes?
The primary beta thalassemia causes are mutations in the HBB gene on chromosome 11. These mutations impair the body’s ability to produce beta-globin, a necessary component of healthy hemoglobin.
2. How does a genetic mutation in beta thalassemia affect a person?
A genetic mutation in beta thalassemia prevents red blood cells from maturing properly. This results in small, fragile red blood cells that die quickly, leading to severe anemia and oxygen deprivation in tissues.
3. Are all inherited blood disorders fatal?
No. Inherited blood disorders range widely in severity. While conditions like Beta Thalassemia major require intensive, lifelong treatment, carrier states like Beta Thalassemia minor usually cause no major health problems.
4. What happens when hemoglobin gene defects occur?
Hemoglobin gene defects cause the body to produce abnormal hemoglobin. This triggers chronic anemia, forces the bone marrow to overwork, and leads to secondary complications like bone deformities and dangerous iron accumulation.
5. Why do beta thalassemia patients need platelet transfusions?
Beta thalassemia causes enlarged spleens and bone marrow suppression, which destroy and limit the production of platelets. Platelet transfusions are necessary to prevent or stop life-threatening bleeding when platelet counts drop dangerously low.
6. How long does a platelet transfusion take?
A platelet transfusion is a relatively quick procedure. The actual intravenous infusion typically takes between 15 to 30 minutes, though pre-transfusion matching and post-transfusion monitoring add to the total hospital time.
7. Can platelet transfusions cure beta thalassemia?
No. Platelet transfusions only manage the symptom of severe thrombocytopenia by temporarily restoring clotting ability. They do not fix the underlying genetic defect or cure the disease.
8. What is alloimmunization in the context of transfusions?
Alloimmunization occurs when a patient’s immune system recognizes transfused donor platelets as foreign invaders and creates antibodies against them. This makes future transfusions much less effective because the immune system destroys the new platelets immediately.
9. Why is iron chelation therapy important?
Patients receiving regular red blood cell transfusions accumulate toxic levels of iron in their organs. Iron chelation therapy uses medications to bind and remove this excess iron, preventing fatal heart and liver failure.
10. Should all beta thalassemia patients have their spleen removed?
No. Doctors only recommend a splenectomy if the spleen becomes massively enlarged and starts destroying too many red blood cells and platelets. Removing the spleen increases the patient’s lifetime risk of severe bacterial infections.