


Most people who carry the beta thalassemia trait have no idea. They feel fine. Their energy levels are normal. Their daily lives are completely unaffected. Yet hidden inside their DNA is a genetic variation that could have profound implications—not for their own health, but for the health of their children.
Beta thalassemia trait is one of the most common inherited blood disorders in the world, particularly prevalent across the Mediterranean, Middle East, South Asia, and Southeast Asia. According to the World Health Organization (WHO), approximately 1.5% of the global population are carriers of some form of beta-thalassemia mutation. That translates to millions of people walking through life with this genetic status, most of them completely unaware.
Understanding the beta thalassemia trait is more than a medical exercise. It is a matter of family planning, reproductive decision-making, and—most importantly—preventing one of the most severe forms of inherited blood disease. When two carriers have children together, there is a 25% chance that each pregnancy will result in a child with beta thalassemia major, a life-threatening condition requiring lifelong blood transfusions and intensive medical care.
This comprehensive guide covers everything carriers and prospective parents need to know: what beta thalassemia trait actually is at a genetic level, how it differs from the disease itself, what symptoms (if any) to expect, how it is diagnosed, how to live well with it, and what steps to take during pregnancy. The science is clear, the stakes are real, and the knowledge available today can make a fundamental difference.
What Is Beta Thalassemia Trait and How Does It Differ from the Disease?
 Beta thalassemia trait—sometimes called beta thalassemia minor—is the carrier state of beta thalassemia. A person with this condition has inherited one normal beta-globin gene and one mutated beta-globin gene. The single functioning gene is enough to maintain near-normal hemoglobin production, which is why most carriers experience few or no symptoms throughout their lives.
Beta thalassemia trait—sometimes called beta thalassemia minor—is the carrier state of beta thalassemia. A person with this condition has inherited one normal beta-globin gene and one mutated beta-globin gene. The single functioning gene is enough to maintain near-normal hemoglobin production, which is why most carriers experience few or no symptoms throughout their lives.
Beta thalassemia as a disease exists on a spectrum. At the mild end sits the trait (minor). In the middle is beta thalassemia intermedia, where anemia is moderate and may occasionally require transfusions. At the severe end is beta thalassemia major—also known as Cooley’s anemia—where both beta-globin genes are defective, hemoglobin production is severely impaired, and regular blood transfusions are required for survival. Understanding where the trait falls on this spectrum is crucial. Being a carrier is not a disease. It is simply a genetic status with meaningful implications for family planning.
To understand the distinction more fully, consider how the beta-globin gene functions in normal hemoglobin production, and what happens when one copy carries a mutation.
The Genetics Behind Beta Thalassemia Trait
What Is Hemoglobin and Why Does Beta-Globin Matter?
Hemoglobin is the iron-containing protein inside red blood cells responsible for carrying oxygen from the lungs to every tissue in the body. Normal adult hemoglobin (HbA) is made up of two alpha-globin chains and two beta-globin chains. These four chains work in precise balance. When beta-globin production is reduced or absent, the balance breaks down, leading to ineffective red blood cell production and varying degrees of anemia.
The instructions for making beta-globin come from the HBB gene on chromosome 11. More than 300 mutations in this gene have been identified as causes of beta thalassemia. Some mutations reduce beta-globin production (called beta+ mutations), while others eliminate it entirely (called beta0 mutations). The specific mutation a carrier has inherited influences how severely the condition may express in offspring who inherit two mutated copies.
How Autosomal Recessive Inheritance Works in Beta Thalassemia
Beta thalassemia follows an autosomal recessive inheritance pattern. Each parent has two copies of the HBB gene—one inherited from each grandparent. A carrier (a person with beta thalassemia trait) has one normal copy and one mutated copy.
When two carriers have children, each pregnancy has four possible genetic outcomes:
- 25% chance: child inherits two normal genes (unaffected, not a carrier)
- 50% chance: child inherits one normal and one mutated gene (carrier, like the parents)
- 25% chance: child inherits two mutated genes (beta thalassemia major or intermedia, depending on mutation type)
This 1-in-4 risk per pregnancy is why genetic counseling is so important for couples who are both carriers of the beta thalassemia trait. For a broader understanding of how related blood conditions compare, read our guide on thalassemia chipmunk face, which illustrates the severe bone changes that can occur in untreated beta thalassemia major.
How Is Beta Thalassemia Trait Different from Beta Thalassemia Major?
The table below summarizes the key differences:
|
Feature |
Beta Thalassemia Trait |
Beta Thalassemia Major |
|---|---|---|
|
Gene copies affected |
One (heterozygous) |
Both (homozygous) |
|
Symptoms |
Mild or none |
Severe |
|
Anemia |
Mild microcytic |
Severe, life-threatening |
|
Treatment required |
Rarely |
Lifelong transfusions |
|
Life expectancy |
Normal |
Reduced without treatment |
Understanding this distinction helps carriers avoid unnecessary anxiety while remaining appropriately informed about the implications for their children. For a detailed look at long-term outcomes in severe thalassemia, see our article on beta thalassemia life expectancy.
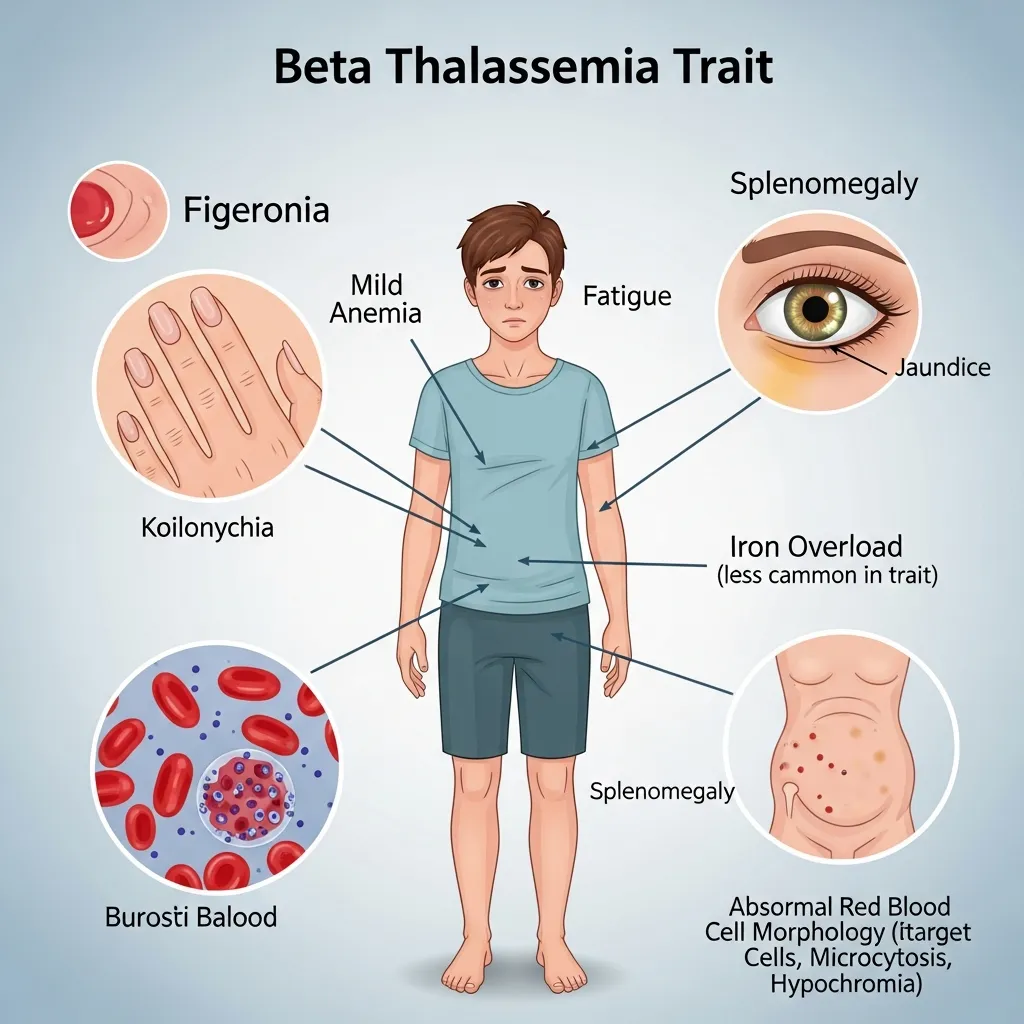
Symptoms and Clinical Presentation of Beta Thalassemia Trait
Why Are Most Carriers Asymptomatic?
The single functional beta-globin gene produces enough beta-globin to support hemoglobin synthesis at near-normal levels. As a result, the body can maintain sufficient oxygen delivery to tissues, and most carriers live entirely symptom-free. This is both a relief and a risk—because the absence of symptoms means many carriers never seek testing and remain unaware of their genetic status.
Mild Anemia: What It Looks Like
Some carriers do develop mild anemia, typically discovered incidentally during routine blood testing. This anemia is usually microcytic (small red blood cells) and hypochromic (pale red blood cells), reflecting slightly reduced hemoglobin content. In practical terms, a carrier might experience:
- Occasional fatigue, particularly after physical exertion
- Mild pallor
- Slight weakness during periods of increased physiological demand, such as illness or pregnancy
These symptoms are generally manageable and do not require medical intervention in most cases. They can, however, be easily mistaken for iron deficiency anemia—a distinction that carries important consequences, as we will explore in the diagnosis section.
Rare Clinical Features
Splenomegaly (an enlarged spleen) and jaundice are occasionally seen in some carriers, though they are far more characteristic of more severe thalassemia forms. When these features appear in a carrier, they usually indicate an additional co-existing condition or a more complex genetic profile and warrant further investigation.
How Is Beta Thalassemia Trait Diagnosed?
Diagnosis requires specific laboratory testing. A routine physical exam will rarely reveal a carrier, and even standard blood tests need proper interpretation. The diagnostic workup typically involves several steps.
Complete Blood Count (CBC): The First Clue
The CBC is usually the starting point. In carriers, characteristic findings include:
- Microcytosis: Smaller-than-normal red blood cells (low Mean Corpuscular Volume, or MCV, typically below 80 fL)
- Hypochromia: Red blood cells with lower-than-normal hemoglobin content
- Elevated red blood cell count (RBC): Despite small cell size, the total number of red blood cells is often high or at the upper limit of normal—a key distinguishing feature from iron deficiency anemia, where RBC count is typically low
This combination—low MCV with a high or normal RBC count—raises a strong clinical suspicion for beta thalassemia trait.
Hemoglobin Electrophoresis and HPLC: Confirming the Diagnosis
Hemoglobin electrophoresis and High-Performance Liquid Chromatography (HPLC) are the definitive tests for identifying carrier status. These techniques separate the different types of hemoglobin present in the blood. In beta thalassemia trait, the hallmark finding is:
- Elevated HbA2 level: HbA2 (a minor form of adult hemoglobin) is typically above 3.5%, compared to the normal range of 1.5–3.5%
- Mildly elevated HbF (fetal hemoglobin): Some carriers show a slight increase in HbF, though this is not always present
An elevated HbA2 is considered the most reliable biochemical marker for beta thalassemia trait. The hemoglobin panel is the essential test for any person with a family history of thalassemia or unexplained microcytic anemia.
Genetic Testing: The Gold Standard for Confirmation
While HPLC confirms carrier status in most cases, DNA analysis of the HBB gene provides the most precise diagnostic information. Genetic testing:
- Identifies the exact mutation(s) present
- Predicts the potential severity of disease in offspring (important when both parents are carriers)
- Allows for accurate prenatal testing
- Helps distinguish carriers of beta+ mutations from those with beta0 mutations
According to the Centers for Disease Control and Prevention (CDC), genetic testing is particularly important for reproductive planning and should be offered to any couple where both partners are identified as carriers.
Why Distinguishing Beta Thalassemia Trait from Iron Deficiency Anemia Matters
Both conditions produce microcytic, hypochromic anemia. Misdiagnosing beta thalassemia trait as iron deficiency anemia—and prescribing iron supplements as a result—is one of the most common clinical errors. Here is why it matters:
|
Feature |
Beta Thalassemia Trait |
Iron Deficiency Anemia |
|---|---|---|
|
RBC count |
High or normal |
Low |
|
HbA2 level |
Elevated (>3.5%) |
Normal or low |
|
Serum ferritin |
Normal |
Low |
|
Response to iron |
No improvement |
Significant improvement |
Iron supplementation in a carrier without iron deficiency carries a risk of iron overload over time, particularly when combined with other factors. Accurate testing avoids this entirely preventable complication.
Management and Living with Beta Thalassemia Trait

No Specific Treatment Is Usually Required
The most important thing for most carriers to understand is this: beta thalassemia trait does not require treatment in the vast majority of cases. The mild anemia associated with the trait rarely causes significant symptoms and does not progress to a more severe condition over time. There is no medication to “correct” carrier status, nor is there any need for one.
A diagnosis of beta thalassemia trait should not change a person’s daily life in any meaningful physical way. What it should change is their awareness of reproductive risks—and prompt a conversation with a healthcare provider about family planning.
Folic Acid Supplementation
Some clinicians recommend low-dose folic acid supplementation for carriers, particularly during periods of increased physiological demand such as pregnancy or illness. Folic acid supports healthy red blood cell production and may help maintain hemoglobin levels during these times. However, routine supplementation in asymptomatic carriers outside of pregnancy is a matter of clinical discretion rather than a universal recommendation.
Avoiding Unnecessary Iron Supplementation
Unless iron deficiency has been confirmed through blood testing (low serum ferritin, low transferrin saturation), carriers should not take iron supplements. Iron-rich diets and routine iron supplementation are not harmful for the general population, but they can be unnecessarily harmful for carriers who are not iron-deficient.
Regular Medical Check-Ups
Carriers benefit from periodic blood count monitoring, particularly during major physiological changes such as pregnancy, significant illness, or when additional stressors arise. Annual check-ups that include a CBC are reasonable for most carriers. This also provides an ongoing opportunity for healthcare providers to revisit family planning discussions and ensure carriers have the most current information.
Lifestyle Adjustments and Diet
Most lifestyle recommendations for carriers center on general good health rather than disease-specific management:
- Balanced diet: Focus on foods rich in folate (leafy greens, legumes, citrus) to support red blood cell production
- Adequate hydration: Helps maintain healthy blood viscosity
- Regular exercise: Cardiovascular fitness supports oxygen delivery efficiency and overall well-being
- Stress management: Chronic stress can exacerbate fatigue, a mild symptom some carriers experience
For those managing mild fatigue, iron-rich foods are appropriate only if iron deficiency has been separately confirmed by blood testing. For comprehensive dietary guidance for those with more complex thalassemia conditions, the site’s thalassemia diet and nutrition resource provides detailed recommendations.
Beta Thalassemia Trait and Pregnancy
Why Pregnancy Requires Special Attention for Carriers
Pregnancy places significant demands on the body’s blood-forming system. The blood volume expands by approximately 40–50% during pregnancy, and the demand for hemoglobin production increases substantially. For most carriers, the body adapts adequately, though hemoglobin levels may decline slightly below the normal pregnancy range—making regular monitoring essential.
Anemia during pregnancy, even mild anemia, increases the risk of fatigue, reduced fetal oxygen delivery, and pregnancy complications. Carriers should inform their obstetric team of their status at the outset of pregnancy so that appropriate monitoring can be arranged.
Risk of Beta Thalassemia Major in Offspring: What Couples Need to Know
The central concern in pregnancy for a carrier is not the carrier’s own health, but the genetic risk to their child. If the partner is also a carrier of beta thalassemia trait—or carries another significant beta-globin mutation—each pregnancy carries a 25% chance of resulting in a child with beta thalassemia major.
This risk is real and well-documented. According to WHO estimates, approximately 56,000 children are born each year with severe thalassemia globally. A significant proportion of these births occur to parents who were carriers but had not received genetic counseling or carrier testing before conception.
For context on how alpha thalassemia carrier status interacts with beta thalassemia in reproductive planning, see our article on alpha thalassemia carrier pregnancy.
Genetic Counseling for Couples: The Essential Step
When one partner is identified as a carrier of beta thalassemia trait, partner testing is the logical and important next step. If the partner is not a carrier, the children cannot have beta thalassemia major (they may be carriers themselves, but they will not develop the severe disease). If the partner is also a carrier, genetic counseling provides the couple with clear, evidence-based information about:
- The 25% risk per pregnancy of beta thalassemia major
- The 50% probability of having a carrier child
- The 25% chance of having a completely unaffected child
- Prenatal diagnostic options available to them
- Psychological and emotional support resources
A genetic counselor does not make decisions for families. They provide information and support so that couples can make informed choices aligned with their values.
Prenatal Diagnosis Options: CVS and Amniocentesis
For couples who are both carriers, two established prenatal diagnostic options exist:
Chorionic Villus Sampling (CVS): Performed at 10–13 weeks of pregnancy, CVS involves taking a small sample of placental tissue for genetic analysis. It can determine whether the fetus has inherited two mutated beta-globin genes (beta thalassemia major), one mutated gene (carrier), or two normal genes. CVS carries a small procedural risk of miscarriage, estimated at approximately 0.5–1%.
Amniocentesis: Performed at 15–20 weeks, amniocentesis involves sampling the amniotic fluid surrounding the fetus. It provides the same genetic information as CVS but is performed later in pregnancy. The miscarriage risk is slightly lower, around 0.1–0.5%.
Both procedures are performed under ultrasound guidance and provide definitive genetic information. Couples should discuss both options thoroughly with their obstetrician and genetic counselor before making a decision.
Managing Pregnancy in a Carrier
For a carrier whose partner is not a carrier, pregnancy management is largely routine with the following additions:
- Monitor hemoglobin and hematocrit at each prenatal visit
- Ensure iron supplementation is only prescribed if iron deficiency is confirmed separately
- Prescribe folic acid supplementation (standard in all pregnancies, and particularly relevant for carriers)
- Educate the patient that mild anemia may develop or worsen in the third trimester and is not cause for alarm if adequately monitored
For carriers whose partner is also a carrier, prenatal diagnosis decisions should ideally be made before 13 weeks of pregnancy to allow for CVS if desired.
Myths and Misconceptions About Beta Thalassemia Trait

Myth 1: Beta Thalassemia Trait Is the Same as Beta Thalassemia Disease
This is perhaps the most damaging misconception. The trait is a carrier state, not a disease. A person with the trait has one functional gene producing sufficient hemoglobin for normal or near-normal health. Beta thalassemia major—the disease—requires two defective genes and causes severe, life-threatening anemia that demands lifelong medical management. Conflating the two causes unnecessary anxiety in carriers and may even lead some to avoid disclosing their status out of fear of stigma.
Myth 2: Carriers Cannot Exercise, Work, or Live Normally
Completely false. The overwhelming majority of people with beta thalassemia trait live fully active, healthy lives with no physical restrictions whatsoever. Athletes, professionals, and individuals across all walks of life carry this trait without any impact on performance or longevity. The only meaningful lifestyle implication is in the context of family planning.
Myth 3: Beta Thalassemia Trait Can Be “Caught” from Another Person
Thalassemia is a genetic condition—entirely inherited, not contagious. No form of contact, shared environment, or exposure can transmit the trait from one person to another. It is passed exclusively through genes from parent to child.
Myth 4: If You Are a Carrier, Your Children Will Definitely Have the Disease
This is incorrect. A carrier’s children will only be at risk of beta thalassemia major if the other parent is also a carrier (or carries another significant beta-globin mutation). If the other parent has two normal beta-globin genes, the children can be either unaffected or carriers themselves, but they cannot develop beta thalassemia major.
Myth 5: Iron Supplements Will Help the Anemia in Beta Thalassemia Trait
As discussed in the diagnosis section, the mild anemia in beta thalassemia trait is caused by reduced beta-globin production—not iron deficiency. Iron supplements will not improve this anemia and may cause harm if taken long-term without confirmed iron deficiency. Always test serum ferritin before prescribing or taking iron supplements in a carrier.
Research and Future Directions in Beta Thalassemia
Advances in Genetic Counseling and Carrier Screening
Carrier screening programs have dramatically reduced the incidence of beta thalassemia major in several countries. Cyprus, for example, implemented a national screening program in the 1970s and reduced the number of new beta thalassemia major births from approximately 14 per year to near zero. Greece, Italy, and Iran have achieved similar results through national screening and genetic counseling initiatives. These programs demonstrate clearly that identifying carriers before reproduction, combined with accurate counseling and access to prenatal diagnosis, is the most effective public health strategy for reducing the burden of severe thalassemia.
Advances in next-generation sequencing (NGS) are making genetic testing faster, more affordable, and more accessible globally. As the cost of comprehensive genetic panels continues to decline, carrier screening before marriage or conception is increasingly feasible even in lower-resource settings.
Potential Therapeutic Interventions: Gene Therapy and the Future
While gene therapy is primarily being developed for patients with beta thalassemia major rather than carriers, the progress in this field has significant implications. Gene therapy approaches—including lentiviral vector gene addition and CRISPR-based gene editing—are already showing promising clinical results in patients with severe beta thalassemia. For a detailed look at these developments, see our article on gene therapy for thalassemia.
For carriers, the most immediately relevant future development is not treatment but rather expanded access to preimplantation genetic testing (PGT), which allows embryos created through IVF to be screened for beta thalassemia mutations before implantation. This technology already exists and is available in many countries, giving carrier couples the option to select unaffected embryos.
Global Awareness Campaigns
Organizations including the Thalassemia International Federation (TIF), the World Health Organization (WHO), and national health ministries in high-prevalence regions continue to expand awareness campaigns targeting young adults before they enter reproductive age. The Thalassemia International Federation coordinates global efforts to improve access to screening, counseling, and treatment for affected individuals and families worldwide.
The goal of awareness is not fear—it is empowerment. Knowing your carrier status before you plan a family is one of the most powerful pieces of health information a person can have.
Empowering Carriers: Knowledge Is the Most Effective Tool
 A diagnosis of beta thalassemia trait should not be a source of fear or stigma. The vast majority of carriers live completely healthy, normal lives. The physical impact of carrying one mutated beta-globin gene is minimal. The social and reproductive impact, however, depends entirely on awareness and access to information.
A diagnosis of beta thalassemia trait should not be a source of fear or stigma. The vast majority of carriers live completely healthy, normal lives. The physical impact of carrying one mutated beta-globin gene is minimal. The social and reproductive impact, however, depends entirely on awareness and access to information.
Carriers who know their status can:
- Ensure their partner is tested before planning a family
- Access genetic counseling to understand their specific risks
- Make informed reproductive decisions with full knowledge of available options
- Avoid unnecessary, potentially harmful treatments like unsupervised iron supplementation
- Become advocates within their families, encouraging siblings and parents to be tested
The countries with the lowest rates of severe thalassemia births are those where carrier screening is normalized and accessible. Every carrier who understands their status and communicates it within their family network helps break the cycle of uninformed reproduction that leads to preventable cases of beta thalassemia major.
For those navigating these conversations and decisions, support groups, patient advocacy organizations, and healthcare providers specializing in hemoglobin disorders are valuable resources. Understanding related conditions—like the impact of hemophilia and low platelet count in other inherited blood disorders—can also help place the beta thalassemia trait within the broader landscape of genetic blood conditions.
Beta Thalassemia Trait: What You Need to Know and Do
Being a carrier of the beta thalassemia trait does not define your health—but it does shape the health decisions you may need to make for your family. Here are the practical takeaways:
- Get tested if you have a family history of thalassemia, or if you are from a high-prevalence region (Mediterranean, Middle East, South Asia, Southeast Asia, or Sub-Saharan Africa)
- Confirm diagnosis with HPLC or hemoglobin electrophoresis, not just a CBC
- Ensure your partner is tested before planning a pregnancy
- Seek genetic counseling if both you and your partner are carriers
- Do not take iron supplements unless iron deficiency has been separately confirmed by blood testing
- Inform your obstetric team of your carrier status at the start of pregnancy
- Consider prenatal diagnosis if both partners are carriers and wish to know the genetic status of the fetus
Beta thalassemia trait is not a disease. But it carries knowledge—and that knowledge, used wisely, can prevent significant suffering in the next generation. Take the appropriate steps, have the right conversations, and work with a healthcare provider who understands hemoglobin disorders. The resources are available. The decisions are yours to make.
Conclusion
Beta Thalassemia Trait is a mild genetic carrier condition that usually does not cause serious health problems, and most individuals live normal lives without treatment. In some cases, it may lead to mild anemia or slight fatigue, but symptoms are generally minimal. The key importance of this condition lies in its genetic nature rather than its clinical impact. Identifying the Beta Thalassemia Trait is essential for understanding carrier status and preventing the transmission of severe thalassemia. Genetic counseling plays a vital role in helping individuals and families make informed reproductive decisions. Early screening allows couples to assess risks before planning a family. Awareness of carrier status also supports public health efforts to reduce the incidence of inherited blood disorders. Although treatment is not required, regular health check-ups can help monitor overall well-being. Education about the Beta Thalassemia Trait helps reduce confusion and stigma surrounding the condition. Overall, early detection and awareness are key to preventing future generations from being affected by severe thalassemia.
Frequently Asked Questions About Beta Thalassemia Trait
1. What is the difference between beta thalassemia trait and beta thalassemia major?
Beta thalassemia trait (minor) means a person has inherited one mutated and one normal beta-globin gene. Carriers are usually asymptomatic or have mild anemia and do not require treatment. Beta thalassemia major occurs when both beta-globin genes are defective, causing severe, life-threatening anemia that requires lifelong blood transfusions and specialized medical management.
2. Can a person with beta thalassemia trait pass it to their children?
Yes. A carrier will pass either the normal or mutated beta-globin gene to each child. If the other parent also carries the trait, each pregnancy has a 25% chance of resulting in a child with beta thalassemia major, a 50% chance of producing another carrier, and a 25% chance of an unaffected child.
3. Does beta thalassemia trait require any treatment?
In most cases, no. The vast majority of carriers live normal, healthy lives without any medical intervention. Folic acid supplementation may occasionally be recommended, particularly during pregnancy. Iron supplementation is only appropriate if iron deficiency is separately confirmed by blood testing.
4. How is beta thalassemia trait diagnosed accurately?
Diagnosis involves a Complete Blood Count (CBC) showing microcytosis and elevated RBC count, followed by hemoglobin electrophoresis or HPLC, which reveals elevated HbA2 levels (above 3.5%). Genetic testing of the HBB gene provides the most precise confirmation and identifies the specific mutation.
5. Why is beta thalassemia trait often confused with iron deficiency anemia?
Both conditions produce microcytic, hypochromic anemia on a CBC. The key distinguishing features are: a high or normal RBC count and elevated HbA2 in beta thalassemia trait, versus a low RBC count, low serum ferritin, and normal HbA2 in iron deficiency anemia. Misdiagnosis can lead to inappropriate iron supplementation.
6. What are the prenatal testing options for carrier couples?
Two main options exist: Chorionic Villus Sampling (CVS) at 10–13 weeks of pregnancy and amniocentesis at 15–20 weeks. Both involve sampling fetal genetic material to determine whether the baby has inherited two mutated beta-globin genes. Preimplantation Genetic Testing (PGT) via IVF is another option that screens embryos before implantation.
7. Can a person with beta thalassemia trait exercise and live a normal life?
Yes, completely. The vast majority of carriers have no physical restrictions and can participate in all activities, including intense exercise and demanding professions. The trait has minimal impact on daily physical health for most people.
8. Is carrier screening for beta thalassemia recommended before marriage?
In high-prevalence regions, many healthcare systems and public health programs strongly recommend carrier screening for both partners before or early in pregnancy. Early identification allows couples to understand their risks and access genetic counseling before reproductive decisions are made.
9. How does beta thalassemia trait affect pregnancy?
Most carriers tolerate pregnancy well. Hemoglobin levels may decline slightly more than in non-carriers due to increased physiological demands. Regular monitoring, folic acid supplementation, and informing the obstetric team of carrier status are the primary management steps. If the partner is also a carrier, prenatal diagnosis should be discussed early.
10. What is the global prevalence of beta thalassemia trait?
According to the World Health Organization (WHO), approximately 1.5% of the global population are carriers of beta-thalassemia mutations. Prevalence is highest in the Mediterranean, Middle East, South Asia, Southeast Asia, and parts of Sub-Saharan Africa—regions where malaria has historically been endemic and where the thalassemia mutation may have offered some survival advantage against severe malaria.