


Thalassemia molecular pathology explores how inherited mutations in hemoglobin genes disrupt red blood cell production, leading to anemia and related complications. Understanding these molecular mechanisms improves diagnosis, genetic counseling, treatment planning, and supports advances in targeted therapies and patient care.
Thalassemia molecular pathology studies how mutations in the alpha and beta globin genes reduce hemoglobin production, cause globin chain imbalance, and drive anemia, guiding diagnosis and treatment.
Thalassemia is one of the most common inherited blood disorders in the world, affecting millions of people across the Mediterranean, the Middle East, South Asia, and Southeast Asia. At its core, thalassemia is a problem of broken instructions. Tiny changes in the genes that build hemoglobin throw off the careful balance red blood cells need to carry oxygen.
Thalassemia molecular pathology is the field that explains exactly how those genetic changes lead to disease. It connects a single DNA mutation to the clumping of unpaired globin chains, the early death of red blood cells, and the chronic anemia that patients live with. Understanding this chain of events is the key to accurate diagnosis, smart treatment choices, and better family planning.
This guide walks through the molecular basis of thalassemia from start to finish. You will learn how hemoglobin is built, which genes control it, and how specific hemoglobin gene mutations cause alpha and beta thalassemia. We will also cover the lab techniques used to find these mutations, how genotype shapes clinical severity, and where treatment is heading. By the end, you will have a clear, complete picture of thalassemia pathophysiology at the molecular level.
What Is Thalassemia and Why Does Molecular Pathology Matter?
 Thalassemia is a group of inherited disorders marked by reduced or absent production of one of the globin chains that make up hemoglobin. The result is less functional hemoglobin, fewer healthy red blood cells, and varying degrees of anemia.
Thalassemia is a group of inherited disorders marked by reduced or absent production of one of the globin chains that make up hemoglobin. The result is less functional hemoglobin, fewer healthy red blood cells, and varying degrees of anemia.
The World Health Organization estimates that about 5% of the global population carries a significant hemoglobin variant, and tens of thousands of severely affected children are born each year. This makes thalassemia a major public health concern, especially in regions where carrier rates run high.
Molecular pathology matters because thalassemia is, at its heart, a genetic disease. Two patients with the same diagnosis on a blood test can carry very different mutations and face very different futures. Identifying the exact defect allows doctors to predict severity, offer prenatal testing, match patients to the right treatment, and counsel families about inheritance risk. In short, the molecular basis of thalassemia shapes nearly every clinical decision.
What Is the Molecular Basis of Thalassemia?
To understand thalassemia pathophysiology, you first need to understand hemoglobin and the genes that build it.
Hemoglobin Structure and Function
Hemoglobin is the oxygen-carrying protein packed inside red blood cells. Each molecule has a four-part design: two pairs of globin chains, each cradling a heme group that holds an iron atom. That iron atom is the exact spot where oxygen binds.
Adult hemoglobin (HbA) is made of two alpha-globin chains and two beta-globin chains. These chains must be produced in nearly equal amounts for hemoglobin to form correctly. When that balance breaks, red blood cells suffer. You can explore this partnership in more detail in this guide on the alpha vs beta globin function difference.
The heme group is just as important as the protein chains. Each hemoglobin molecule carries four heme groups, so it can bind up to four oxygen molecules at once. To learn how this loading and unloading works, see this breakdown of hemoglobin structure and oxygen binding.
The Genetic Architecture of Globin Genes
The instructions for globin chains sit in two gene clusters on two different chromosomes.
- Alpha-globin gene cluster (chromosome 16): Each person carries four alpha-globin genes, two inherited from each parent. This four-gene setup explains why alpha-thalassemia comes in graded severity.
- Beta-globin gene cluster (chromosome 11): Each person carries two beta-globin genes, one from each parent. The cluster also holds the genes for gamma, delta, and embryonic chains, arranged in the order they switch on during development.
This genetic layout is central to thalassemia molecular pathology. Because there are four alpha genes but only two beta genes, the two types of thalassemia follow different inheritance patterns and respond differently to hemoglobin gene mutations.
Alpha-Thalassemia: Molecular Defects and Clinical Effects
Alpha-thalassemia results from reduced or absent alpha-globin production. Most cases come from deletions of one or more of the four alpha-globin genes, though point mutations also play a role.
Deletional Alpha-Thalassemia
The severity of deletional alpha-thalassemia tracks directly with the number of genes lost:
- One-gene deletion (silent carrier): Alpha output drops only slightly. These individuals have no symptoms and usually learn of their status only through genetic testing.
- Two-gene deletion (alpha-thalassemia trait): Produces mild anemia and small, pale red blood cells (microcytosis). Symptoms are minimal or absent.
- Three-gene deletion (Hemoglobin H disease): Only one working alpha gene remains. Excess beta chains form unstable Hemoglobin H, leading to moderate to severe anemia and an enlarged spleen.
- Four-gene deletion (Hb Bart’s hydrops fetalis): No alpha chains are made at all. This is a life-threatening condition that usually causes death before or shortly after birth without intervention.
Non-Deletional Alpha-Thalassemia
Not all alpha-thalassemia comes from missing genes. Point mutations in the alpha-globin genes can disrupt how the gene is read, spliced, or translated. These non-deletional defects often destabilize the alpha chain or reduce its production.
Interestingly, some non-deletional mutations cause more severe disease than a simple gene deletion. This molecular variability is one reason genotype testing is so valuable, since two carriers can have very different outcomes.
Beta-Thalassemia: Molecular Defects and Clinical Effects
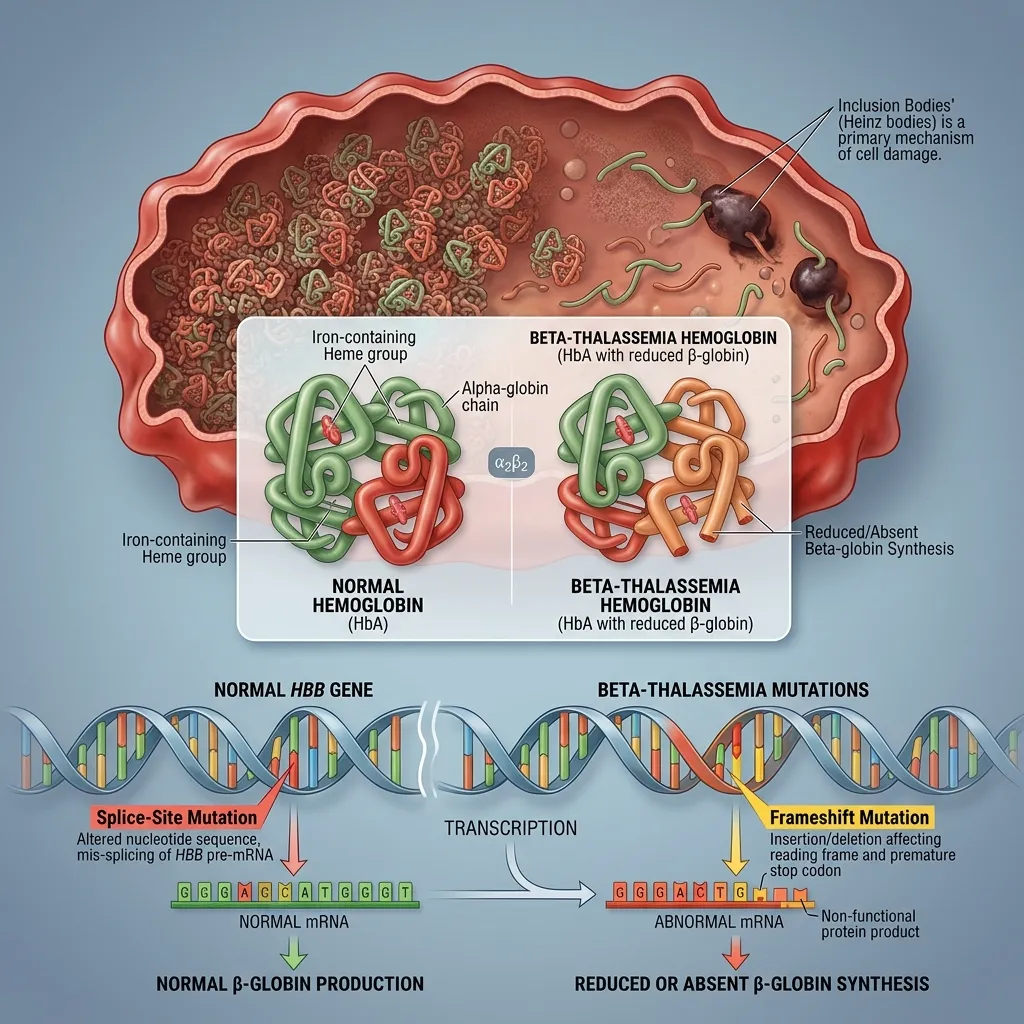 Beta-thalassemia results from mutations in the HBB gene on chromosome 11. Unlike alpha-thalassemia, it is usually caused by point mutations rather than large deletions. More than 300 different HBB mutations have been described, affecting transcription, RNA splicing, and translation.
Beta-thalassemia results from mutations in the HBB gene on chromosome 11. Unlike alpha-thalassemia, it is usually caused by point mutations rather than large deletions. More than 300 different HBB mutations have been described, affecting transcription, RNA splicing, and translation.
These mutations fall into two broad groups. Beta-zero (β⁰) mutations abolish beta-globin production completely, while beta-plus (β⁺) mutations reduce it but leave some output intact. The combination a patient inherits helps determine disease severity.
Beta-Thalassemia Major (Cooley’s Anemia)
Beta-thalassemia major occurs when a person inherits two severe mutations, either as a homozygote or a compound heterozygote. Beta-globin production is severely reduced or absent, so excess alpha chains pile up inside developing red blood cells.
These unpaired alpha chains are toxic. They clump together, damage cell membranes, and trigger the early death of red cell precursors in the bone marrow, a process called ineffective erythropoiesis. The result is severe, transfusion-dependent anemia that appears in early childhood, along with iron overload over time. You can read more about this destructive cycle in this guide on ineffective erythropoiesis.
Beta-Thalassemia Intermedia
Beta-thalassemia intermedia covers a middle ground. It often arises from milder β⁺ mutations or from helpful co-inheritance, such as also carrying alpha-thalassemia, which eases the globin chain imbalance. Patients have moderate anemia and may need only occasional transfusions during illness, surgery, or pregnancy.
Beta-Thalassemia Minor (Trait)
Beta-thalassemia minor results from a single mutated HBB gene. Carriers are usually asymptomatic or have only mild anemia. Blood tests typically show small red blood cells and elevated HbA2. For a closer look at carrier status, see this beta-thalassemia trait carrier guide.
The root problem in every form of beta-thalassemia is the same: too few beta chains and too many leftover alpha chains. This alpha and beta globin chain imbalance is the engine that drives disease severity.
What Are the Other Forms of Thalassemia?
Beyond the two main types, several other variants show how flexible globin genetics can be.
- Delta-beta thalassemia: Both delta and beta chain production are reduced. The body compensates by keeping fetal hemoglobin (HbF) levels higher than normal, which often softens symptoms.
- Hereditary Persistence of Fetal Hemoglobin (HPFH): A benign condition in which HbF production continues into adulthood. When HPFH coexists with beta-thalassemia, the extra fetal hemoglobin can ease the disease. This protective effect is closely tied to the globin gene switching mechanism.
- HbE/beta-thalassemia: Caused by co-inheriting the hemoglobin E variant with a beta-thalassemia mutation. It is one of the most common severe thalassemia syndromes in Southeast Asia, with a wide range of clinical severity.
Which Molecular Techniques Diagnose Thalassemia?
Confirming thalassemia at the DNA level requires a toolbox of molecular methods. The right technique depends on whether you are hunting for deletions or point mutations.
- DNA extraction and PCR amplification: Nearly every test starts here. DNA is isolated from a blood sample, and polymerase chain reaction (PCR) copies the target globin gene regions many times over for analysis.
- Gap-PCR: A specialized PCR designed to detect known deletions. It is the go-to first-line test for common deletional alpha-thalassemia.
- Restriction Fragment Length Polymorphism (RFLP): Uses enzymes that cut DNA at specific sequences. A mutation that changes a cut site alters the fragment pattern, revealing the defect.
- Allele-Specific Oligonucleotide (ASO) hybridization: Uses short DNA probes that bind only to a specific mutation, making it useful for screening common point mutations in a population.
- DNA sequencing: The gold standard for identifying unknown or rare point mutations. Sequencing reads the exact order of bases in the gene.
- Multiplex Ligation-Dependent Probe Amplification (MLPA): Detects large deletions and duplications across the globin clusters, filling a gap that standard PCR can miss.
- Next-Generation Sequencing (NGS): Reads many genes at once and is increasingly used for comprehensive screening. NGS can detect point mutations, deletions, and rare variants in a single run, making it powerful for complex or unexplained cases.
Choose Gap-PCR or MLPA if you suspect deletional alpha-thalassemia, and choose direct sequencing or NGS if you need to identify rare or unknown point mutations.
How Do Genotype-Phenotype Correlations Work in Thalassemia?
 One of the most clinically useful insights from thalassemia molecular pathology is that the specific mutation often predicts disease severity. A β⁰ mutation, which shuts down beta production entirely, tends to cause more severe disease than a mild β⁺ mutation.
One of the most clinically useful insights from thalassemia molecular pathology is that the specific mutation often predicts disease severity. A β⁰ mutation, which shuts down beta production entirely, tends to cause more severe disease than a mild β⁺ mutation.
But genotype is not the whole story. Several modifying factors shape the final clinical picture:
- Co-inheritance of alpha-thalassemia: Reduces excess alpha chains, easing the imbalance and softening beta-thalassemia.
- Elevated fetal hemoglobin: Gamma chains can pair with surplus alpha chains, reducing toxicity. Genetic variants that raise HbF are protective.
- Genetic modifiers: Variations in genes like BCL11A influence how much fetal hemoglobin a person makes, which in turn affects severity.
This is why two patients with identical HBB mutations can still differ in how sick they become. Understanding these modifiers is central to thalassemia pathophysiology and to predicting outcomes.
What Are the Therapeutic Implications of Molecular Pathology?
Pinning down the molecular defect does more than label the disease. It directly shapes treatment and prevention.
Genetic Counseling and Prenatal Diagnosis
Carrier screening and genetic testing let couples understand their risk of having an affected child. Prenatal diagnosis, through chorionic villus sampling or amniocentesis, can identify thalassemia early in pregnancy, giving families informed choices.
Bone Marrow Transplant and Gene Therapy
A hematopoietic stem cell (bone marrow) transplant remains the only established cure for severe thalassemia, working best in young patients with a well-matched donor. Gene therapy is a newer option that corrects or compensates for the faulty gene in a patient’s own stem cells. CRISPR-based approaches that reactivate fetal hemoglobin have shown real promise in reducing transfusion dependence. You can learn more in this overview of gene therapy for thalassemia.
Pharmacological Approaches
Several drugs target the downstream effects of globin chain imbalance:
- Hydroxyurea: Boosts fetal hemoglobin, which can pair with excess alpha chains and ease anemia.
- Iron chelation therapy: Removes the excess iron that builds up from transfusions and increased gut absorption, protecting the heart and liver.
- Newer agents: Drugs that improve red cell maturation can reduce transfusion needs in some patients.
Future Directions
As gene editing matures, base editing and prime editing promise even more precise correction of hemoglobin gene mutations. Combined with cheaper sequencing, these tools point toward truly personalized thalassemia care.
What Challenges Remain in Thalassemia Molecular Research?
 Despite remarkable progress, several hurdles remain.
Despite remarkable progress, several hurdles remain.
- Rare and novel mutations: Hundreds of mutations are known, but new ones keep turning up. Some defy easy classification, complicating diagnosis and counseling.
- Global disparities in diagnostic access: Advanced molecular testing is concentrated in wealthier regions, while many high-prevalence areas lack the resources for comprehensive screening.
- Personalized medicine: Translating genotype data into tailored treatment plans is still evolving. Bridging that gap requires both better data and broader access.
Conclusion
Thalassemia molecular pathology ties a single genetic change to a cascade of consequences: reduced globin production, toxic chain imbalance, ineffective erythropoiesis, and chronic anemia. The molecular basis of thalassemia explains why disease severity ranges from silent carrier to life-threatening, and why two patients with the same diagnosis can follow very different paths.
The good news is that decoding hemoglobin gene mutations has transformed care. Precise molecular diagnosis now guides genetic counseling, prenatal testing, and treatment selection, while gene therapy and HbF-reactivating drugs offer hope for lasting relief.
If you or a family member may be affected by a hemoglobin disorder, the best first step is proper diagnosis through clinical and genetic testing. Speak with a hematology specialist about your options, and keep learning through trusted resources on inherited blood conditions.
Frequently Asked Questions
1. What is thalassemia molecular pathology?
Thalassemia molecular pathology is the study of how mutations in the alpha and beta globin genes disrupt hemoglobin production. It explains how a genetic change leads to globin chain imbalance, ineffective red blood cell production, and anemia, and it guides diagnosis and treatment.
2. What is the molecular basis of thalassemia?
The molecular basis of thalassemia lies in mutations or deletions in the globin genes. Alpha-globin genes sit on chromosome 16, and beta-globin genes sit on chromosome 11. When these genes are faulty, the body cannot make balanced amounts of globin chains, leading to defective hemoglobin.
3. What types of hemoglobin gene mutations cause thalassemia?
Alpha-thalassemia is usually caused by gene deletions, while beta-thalassemia is mostly caused by point mutations affecting transcription, splicing, or translation. Beta-zero mutations stop globin production entirely, and beta-plus mutations reduce it partially.
4. How is thalassemia diagnosed at the molecular level?
Molecular diagnosis uses techniques like PCR, Gap-PCR for deletions, DNA sequencing for point mutations, MLPA for large deletions, and Next-Generation Sequencing for comprehensive screening. The choice depends on whether deletions or point mutations are suspected.
5. Why does beta-thalassemia tend to be more severe than alpha-thalassemia?
In beta-thalassemia, excess alpha chains accumulate and form highly unstable, toxic aggregates that destroy developing red blood cells. In alpha-thalassemia, the excess beta chains are generally less toxic, which often results in milder disease.
6. Can the same mutation cause different disease severity?
Yes. Genetic modifiers strongly influence outcome. Co-inheriting alpha-thalassemia, carrying variants that raise fetal hemoglobin, or other genetic factors can soften the effects of an identical beta-globin mutation.
7. Is thalassemia inherited?
Yes. Thalassemia follows an autosomal recessive pattern. A child must inherit faulty globin genes from both parents to develop a severe form, while inheriting a single mutated gene usually results in carrier status.
8. Can molecular testing help with family planning?
Absolutely. Carrier screening and genetic testing reveal the risk of passing thalassemia to children. Prenatal diagnosis can identify an affected pregnancy early, helping families make informed decisions.
9. How does molecular pathology guide treatment?
Knowing the exact mutation helps doctors predict severity, choose between transfusion strategies, and decide whether a patient is a candidate for a stem cell transplant or gene therapy. It also identifies patients likely to respond to fetal hemoglobin-boosting drugs.
10. What is the future of thalassemia treatment?
The future centers on gene therapy and gene editing, including CRISPR, base editing, and prime editing, which aim to correct hemoglobin gene mutations or reactivate fetal hemoglobin. Combined with cheaper genetic testing, these advances point toward personalized, potentially curative care.