


The Role of Genetic Mutations in Thalassemia
Every case of thalassemia traces back to a mutation in the genes that direct hemoglobin production. These mutations either reduce the amount of globin chains the body makes or stop production entirely. The imbalance damages red blood cells, shortens their lifespan, and forces the bone marrow to work overtime.
Unlike many diseases, thalassemia is never contagious and never acquired through diet or environment. An inherited thalassemia gene mutation is passed strictly from parents to children through their DNA.
Why Understanding the Genetics Matters
Knowing the genetics behind thalassemia matters for one practical reason: it shapes family planning. Two carriers who understand their status can take steps to prevent a severe outcome in their children. Carriers who get tested early avoid years of confusion and misdiagnosis. For a broader view of how these conditions develop, our guide on hemoglobin synthesis disorders explains the wider category of inherited hemoglobin problems.
How Does the Body Make Hemoglobin?
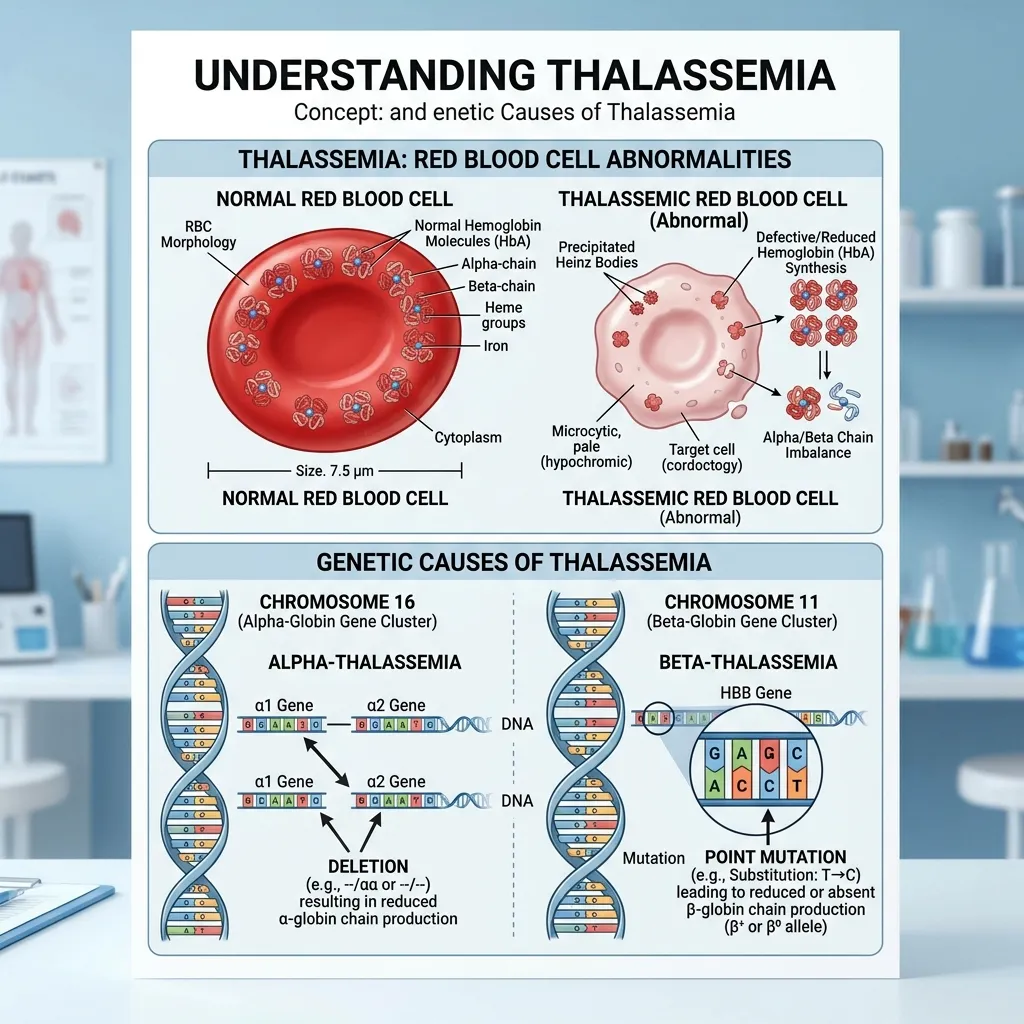
To understand what goes wrong, it helps to know how hemoglobin is built. Normal adult hemoglobin (HbA) consists of two alpha-globin chains and two beta-globin chains working in precise balance.
The instructions for these chains come from specific genes. Alpha-globin production is controlled by genes on chromosome 16, while beta-globin depends on the HBB gene on chromosome 11. When a thalassemia genetic mutation strikes one of these genes, the body either makes too little of a globin chain or produces a faulty version. This imbalance is the core problem in every type of thalassemia.
What Are the Different Types of Genetic Mutations Causing Thalassemia?
Thalassemia splits into two main categories, named for the globin chain affected by the mutation.
Alpha-Thalassemia Mutations
Alpha-thalassemia results from deletions or mutations in the alpha-globin genes on chromosome 16. Most people inherit four alpha-globin genes—two from each parent. The severity depends on how many of these genes are damaged.
- One gene affected: Silent carrier, no symptoms.
- Two genes affected: Alpha-thalassemia trait, with mild or no anemia.
- Three genes affected: Hemoglobin H disease, causing moderate to severe anemia.
- Four genes affected: Hb Barts hydrops fetalis, typically fatal before or shortly after birth.
Beta Thalassemia Genetic Defects
Beta thalassemia genetic defects come from point mutations in the HBB gene on chromosome 11. More than 300 mutations have been identified, which explains the wide range of severity in patients. Some mutations reduce beta-globin output (called beta+ mutations), while others eliminate it completely (called beta0 mutations).
The condition is classified into three forms. Beta-thalassemia major (Cooley’s anemia) requires lifelong blood transfusions. Beta-thalassemia intermedia causes moderate anemia. Beta-thalassemia minor, or trait, usually causes minimal or no symptoms. For a deeper look at the underlying biology, this detailed guide on beta thalassemia causes explains how these gene defects affect red blood cells.
What Is the Thalassemia Gene Inheritance Pattern?
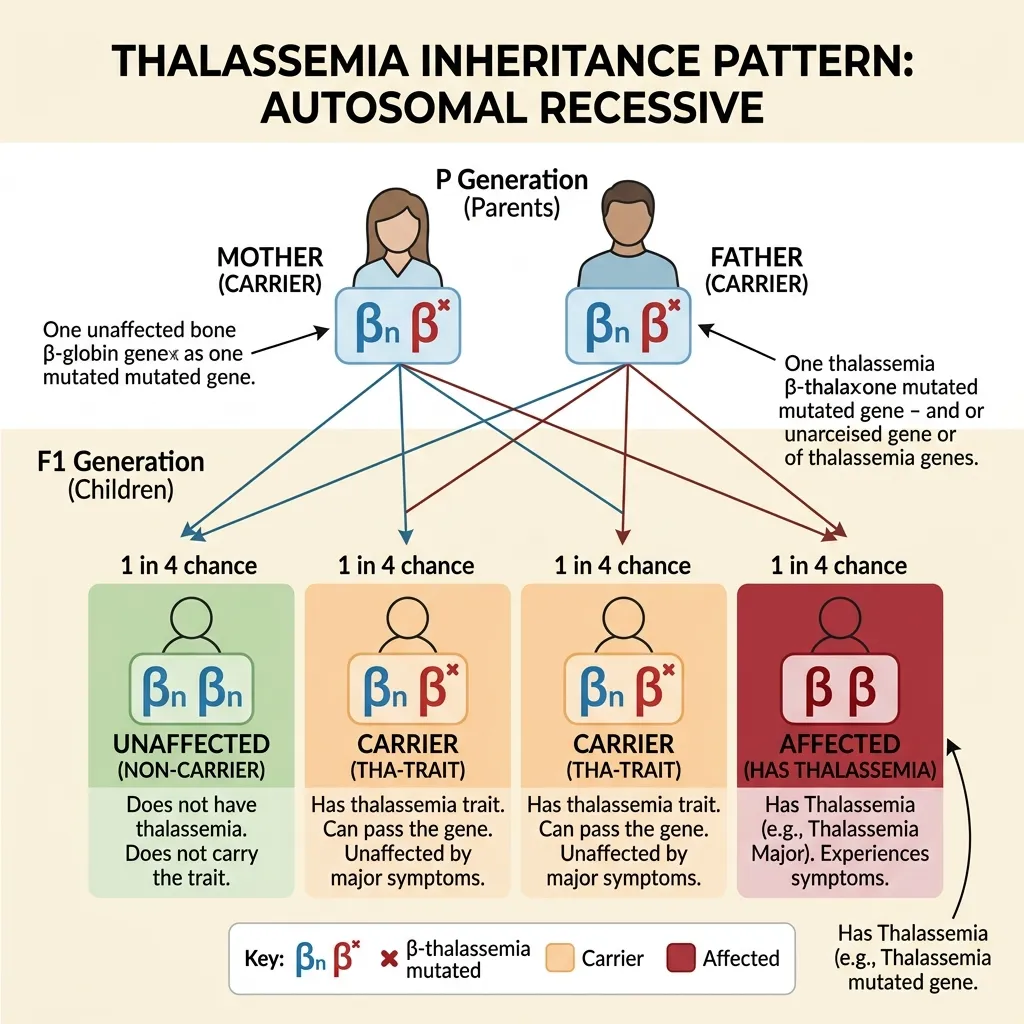
The thalassemia gene inheritance pattern is one of the most important things for families to grasp. It determines who is simply a carrier and who develops the actual disease.
How Autosomal Recessive Inheritance Works
Thalassemia follows an autosomal recessive inheritance pattern. Each person carries two copies of the relevant globin gene—one from each parent. A child must inherit two mutated copies to develop the severe form of the disease.
When two carriers have a child, each pregnancy has four possible outcomes:
- 25% chance: The child inherits two normal genes and is completely unaffected.
- 50% chance: The child inherits one normal and one mutated gene, becoming a carrier like the parents.
- 25% chance: The child inherits two mutated genes and develops beta-thalassemia major or intermedia.
This 1-in-4 risk per pregnancy is exactly why genetic counseling matters so much for carrier couples.
Carriers vs. Affected Individuals
A carrier has one normal gene and one with an inherited thalassemia gene mutation. The single working gene produces enough hemoglobin for near-normal health, which is why most carriers feel completely fine and never know their status.
Affected individuals inherit two mutated genes and cannot make enough functional hemoglobin. This distinction is critical—being a carrier is a genetic status, not a disease. To understand carrier status in depth, our beta thalassemia trait carrier guide offers clear, practical information on diagnosis and family planning.
A Closer Look at Beta-Thalassemia Genetic Defects
Beta-thalassemia deserves special attention because its genetic defects are so varied, and that variation directly shapes how severe the disease becomes.
Specific Gene Loci and Alleles
All beta-thalassemia mutations occur in the HBB gene on chromosome 11. This single gene provides the blueprint for beta-globin. Because so many different mutations can disrupt it, two patients with beta-thalassemia may carry entirely different genetic defects.
Common Mutations and Their Impact
Beta+ mutations reduce beta-globin production but don’t stop it entirely. Patients with these mutations often have milder symptoms. Beta0 mutations, on the other hand, shut down beta-globin production completely, leading to the most severe disease forms. The specific combination a person inherits determines whether they face mild anemia or transfusion-dependent disease.
Genotype-Phenotype Correlation
The link between a person’s exact genetic makeup (genotype) and their actual symptoms (phenotype) is what doctors call genotype-phenotype correlation. Someone who inherits two beta0 mutations typically develops beta-thalassemia major. Someone with one beta+ and one beta0 mutation may land somewhere in the intermedia range. This is why genetic testing—not just a blood count—gives families the clearest picture of what to expect.
When and How Is Thalassemia Genetic Testing Done?
Accurate diagnosis is the foundation of good care, and genetic testing provides the most precise answers.
When Is Genetic Testing Recommended?
Doctors recommend genetic testing in several situations: when a person has a family history of thalassemia, when routine blood work shows unexplained microcytic anemia, when both partners in a couple are identified as carriers, or as part of prenatal and preconception planning. People from high-prevalence regions—the Mediterranean, Middle East, South Asia, and Southeast Asia—are also strong candidates for testing.
Methods of Genetic Testing
Two main laboratory methods identify a thalassemia genetic mutation:
- DNA sequencing: This reads the exact genetic code of the HBB or alpha-globin genes and pinpoints the precise mutation. It’s the gold standard for confirming a diagnosis.
- PCR-based assays: Polymerase chain reaction techniques detect common, known mutations quickly and affordably, making them useful for population screening.
Before genetic testing, doctors usually start with a Complete Blood Count (CBC) and hemoglobin electrophoresis or HPLC. An elevated HbA2 level above 3.5% is a hallmark sign of beta-thalassemia trait.
Interpreting Test Results
Test results identify whether a person carries one or two mutations and which specific defects are present. For carrier couples, this information predicts the potential severity of disease in their children and enables accurate prenatal testing. A genetic counselor helps translate these results into clear, actionable guidance.
How Is Thalassemia Managed and Treated?

Treatment depends heavily on the type and severity of the genetic mutation. For many patients, a combination of approaches works best.
Blood Transfusions
Regular red blood cell transfusions are the cornerstone of treatment for beta-thalassemia major. They maintain healthy hemoglobin levels, suppress ineffective bone marrow activity, and support normal growth in children.
Chelation Therapy
Every transfusion adds iron the body cannot remove on its own. Without intervention, this iron builds up in the heart, liver, and endocrine glands. Iron chelation therapy uses medications that bind to excess iron and help the body excrete it, preventing fatal organ damage.
Bone Marrow Transplant
A bone marrow or stem cell transplant is currently the only established cure for severe thalassemia. The procedure replaces defective marrow with healthy stem cells from a matched donor, usually a sibling. However, it carries significant risks and requires a perfect donor match.
Gene Therapy: A Glimpse into the Future
Gene therapy represents the most exciting frontier in thalassemia treatment. Approaches including lentiviral gene addition and CRISPR-based gene editing are already showing strong clinical results. These techniques correct or work around the faulty gene, offering the potential for a lasting cure without a donor. According to the National Institutes of Health, gene-based therapies continue to advance through clinical trials and offer real hope for transfusion-dependent patients.
How Can a Thalassemia Genetic Mutation Be Prevented?
Prevention focuses on identifying carriers before they have children and giving couples accurate information about their risks.
Pre-implantation Genetic Diagnosis (PGD)
Pre-implantation genetic diagnosis allows embryos created through IVF to be screened for thalassemia mutations before implantation. Carrier couples can select unaffected embryos, dramatically reducing the chance of having a child with severe disease.
Prenatal Diagnosis
For couples who are both carriers, two prenatal tests can determine whether a fetus has inherited two mutated genes. Chorionic villus sampling (CVS) is performed at 10 to 13 weeks, while amniocentesis is done at 15 to 20 weeks. Both provide definitive genetic information under ultrasound guidance.
The Importance of Genetic Counseling for Carriers
Genetic counseling ties everything together. A counselor explains the 25% risk per pregnancy, outlines available options, and provides emotional support—without making decisions for the family. Countries like Cyprus, Greece, and Iran have used national screening and counseling programs to reduce severe thalassemia births to near zero. According to the World Health Organization, carrier screening combined with counseling remains the most effective public health strategy against the disease.
Frequently Asked Questions
What is a thalassemia genetic mutation?
A thalassemia genetic mutation is an inherited change in the genes that produce hemoglobin. It affects either the alpha-globin genes on chromosome 16 or the HBB gene on chromosome 11, reducing the body’s ability to make normal hemoglobin and causing anemia.
What is the thalassemia gene inheritance pattern?
Thalassemia follows an autosomal recessive inheritance pattern. A child must inherit two mutated genes—one from each parent—to develop the severe disease. Inheriting just one mutated gene makes a person a carrier with usually no symptoms.
Can you have a thalassemia mutation and not be sick?
Yes. People who inherit one mutated gene are carriers. Their single working gene produces enough hemoglobin for near-normal health, so most carriers have mild or no symptoms and often don’t know they carry the mutation.
What’s the difference between alpha- and beta-thalassemia mutations?
Alpha-thalassemia mutations affect the alpha-globin genes on chromosome 16, and severity depends on how many of the four genes are damaged. Beta thalassemia genetic defects affect the single HBB gene on chromosome 11, with over 300 known mutations producing a wide range of severity.
How is a thalassemia genetic mutation diagnosed?
Diagnosis usually begins with a Complete Blood Count and hemoglobin electrophoresis or HPLC. DNA sequencing and PCR-based assays then confirm the exact mutation, which helps predict disease severity and guides family planning.
If both parents carry the gene, will their child definitely have thalassemia?
No. When both parents are carriers, each pregnancy carries a 25% chance of an affected child, a 50% chance of a carrier child, and a 25% chance of a completely unaffected child.
Is the thalassemia gene mutation more common in certain groups?
Yes. An inherited thalassemia gene mutation is most common in people of Mediterranean, Middle Eastern, South Asian, and Southeast Asian descent—regions where malaria was historically widespread and carrier status offered some survival advantage.
Can a thalassemia genetic mutation be cured?
A bone marrow or stem cell transplant is currently the only established cure for severe thalassemia. Gene therapy, including CRISPR-based editing, is an emerging option showing strong clinical promise and may offer functional cures without a donor.
Should couples get tested before having children?
Yes. Carrier screening before pregnancy lets couples understand their risks. If both partners carry a thalassemia genetic mutation, genetic counseling and prenatal options like CVS, amniocentesis, or PGD become important next steps.
Does iron supplementation help thalassemia anemia?
No. The anemia in thalassemia comes from reduced globin production, not iron deficiency. Taking iron without a confirmed deficiency can cause harmful iron overload. Always confirm iron levels with a blood test first.
Key Takeaways and Next Steps
A thalassemia genetic mutation may be invisible, but its impact on families is real and lasting. The condition stems from inherited changes in the genes that build hemoglobin, follows a predictable autosomal recessive inheritance pattern, and ranges from a silent carrier state to severe, transfusion-dependent disease.
The path forward comes down to three concrete steps. First, get tested if you have a family history or come from a high-prevalence region. Second, confirm any diagnosis with HPLC or genetic testing rather than relying on a basic blood count. Third, seek genetic counseling before starting a family if you and your partner are both carriers.
Research into gene therapy is rapidly transforming what was once a devastating diagnosis into a condition with genuine hope for a cure. To continue learning, explore our related guides on beta thalassemia causes and hemoglobin synthesis disorders. Early detection and proactive care remain your strongest tools for a healthier future.