


Alpha and Beta Globin Chain Imbalance disrupt normal hemoglobin production, leading to thalassemia and related complications. This guide explains causes, symptoms, diagnosis, treatment options, and management strategies to help patients and caregivers better understand the condition.
Alpha and Beta Globin Chain Imbalance is one of the most important mechanisms behind thalassemia and many hemoglobin disorders. Hemoglobin, the oxygen-carrying protein inside red blood cells, consists of alpha and beta globin chains that must be produced in balanced amounts for healthy red blood cell formation. When the body produces unequal amounts of these chains, normal red blood cell development is disrupted, leading to defective hemoglobin production and impaired oxygen transport throughout the body.
This imbalance causes ineffective red blood cell production, chronic anemia, and a range of complications that vary from mild to life-threatening. Excess globin chains can accumulate inside developing red blood cells, causing cellular damage and premature destruction before the cells mature. As a result, the bone marrow works harder to compensate for the loss, often leading to bone marrow expansion and other long-term complications.
Globin chain imbalance in thalassemia is a key factor that determines disease severity. In alpha thalassemia, reduced alpha chain production leaves excess beta chains, while in beta thalassemia, decreased beta chain production results in harmful accumulation of alpha chains. These alpha and beta globin synthesis defects interfere with the body’s ability to produce stable hemoglobin, creating various thalassemia globin chain abnormalities that affect overall health.
Understanding how Alpha and Beta Globin Chain Imbalance develops helps patients, caregivers, and healthcare professionals better manage thalassemia and improve long-term outcomes. Early diagnosis, appropriate treatment, and regular monitoring can reduce complications, improve quality of life, and support healthier red blood cell production over time. As research advances, a deeper understanding of globin chain balance continues to play a critical role in developing more effective therapies for thalassemia and related blood disorders.
What Are Globin Chains?
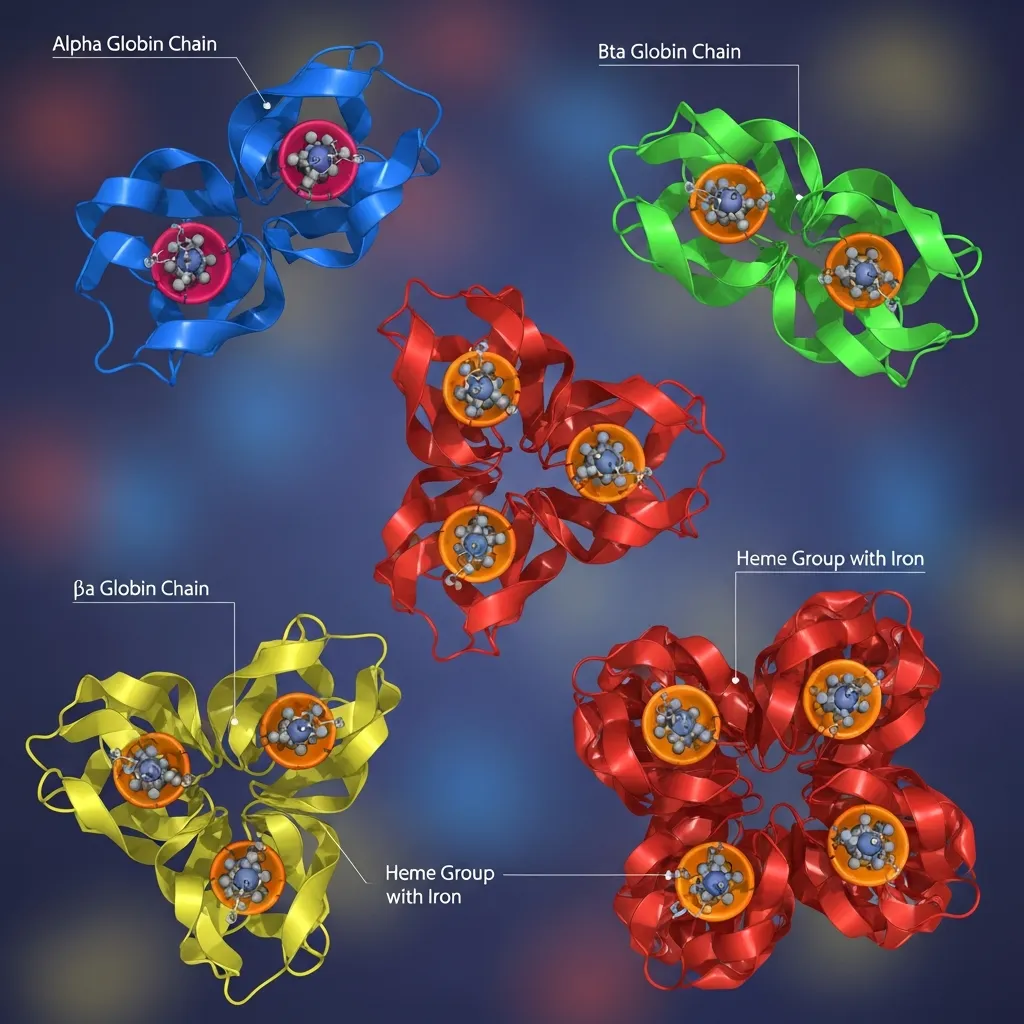 Hemoglobin is the essential protein found inside red blood cells that carries oxygen from the lungs to tissues throughout the body and transports carbon dioxide back to the lungs for removal.
Hemoglobin is the essential protein found inside red blood cells that carries oxygen from the lungs to tissues throughout the body and transports carbon dioxide back to the lungs for removal.
Each hemoglobin molecule is composed of four protein subunits known as globin chains:
- Two alpha globin chains
- Two beta globin chains
These chains work together to form a stable hemoglobin molecule capable of binding and releasing oxygen efficiently. Without the proper structure and balance of these chains, red blood cells cannot function normally, leading to reduced oxygen delivery and various health complications.
The production of globin chains is controlled by specific genes inherited from both parents. The alpha globin genes are located on chromosome 16, while the beta globin genes are located on chromosome 11. These genes provide the instructions needed for the body to manufacture the globin proteins required for healthy hemoglobin formation.
Under normal conditions, alpha and beta chains are produced in carefully regulated amounts. This balanced production ensures that hemoglobin molecules are assembled correctly and that red blood cells remain stable and functional throughout their lifespan. However, when genetic mutations, deletions, or other inherited abnormalities affect either group of genes, the production process becomes disrupted.
As a result, the balance between alpha and beta chain production is disturbed, leading to Alpha and Beta Globin Chain Imbalance. This imbalance is the underlying cause of many forms of thalassemia and other hemoglobin disorders. Depending on which globin chain is affected and the severity of the genetic defect, patients may experience mild anemia, moderate symptoms, or severe life-threatening complications.
Why Is Balance So Important?
The human body relies on a precise balance between alpha and beta globin chain production to create functional hemoglobin. Healthy hemoglobin synthesis requires nearly equal amounts of both types of chains. When this balance is maintained, hemoglobin molecules form correctly, allowing red blood cells to efficiently transport oxygen throughout the body.
Problems arise when one type of globin chain is produced in lower amounts than the other. The excess unmatched chains cannot form normal hemoglobin and instead accumulate within developing red blood cells. These excess chains are often unstable and toxic, causing damage to the cells before they fully mature.
When one globin chain is reduced:
- Excess unmatched chains accumulate.
- Red blood cells become unstable.
- Bone marrow production becomes inefficient.
- Premature destruction of red blood cells occurs.
- Hemoglobin production decreases.
- Oxygen delivery to tissues becomes impaired.
- Chronic anemia develops.
This process contributes directly to the ineffective production of red blood cells, a condition known as ineffective erythropoiesis. Many immature red blood cells die within the bone marrow before entering the bloodstream, forcing the body to work harder to replace them. Over time, this increased demand can lead to bone marrow expansion, skeletal changes, and enlargement of organs such as the spleen and liver.
The severity of disease depends largely on the degree of Globin chain imbalance in thalassemia. Mild imbalances may cause few symptoms, while severe imbalances can result in transfusion-dependent anemia and serious complications. In beta thalassemia, excess alpha chains tend to be highly toxic to developing red blood cells, often leading to more severe disease. In alpha thalassemia, excess beta chains can also cause significant problems, though the clinical presentation may vary depending on the number of affected genes.
Understanding the importance of globin chain balance helps explain why Alpha and beta globin synthesis defects are central to thalassemia pathophysiology. It also highlights the role of Thalassemia globin chain abnormalities in determining disease severity, treatment requirements, and long-term patient outcomes. By maintaining or restoring a better balance of globin chain production, many modern treatment approaches aim to reduce symptoms and improve quality of life for individuals living with thalassemia.
The Role of Hemoglobin in Red Blood Cell Health
 Hemoglobin serves as the body’s primary oxygen transport system and is essential for maintaining healthy body function. Every red blood cell contains millions of hemoglobin molecules that bind oxygen in the lungs and deliver it to tissues and organs throughout the body. This process ensures that cells receive the oxygen needed to produce energy, support growth, and maintain normal physiological functions.
Hemoglobin serves as the body’s primary oxygen transport system and is essential for maintaining healthy body function. Every red blood cell contains millions of hemoglobin molecules that bind oxygen in the lungs and deliver it to tissues and organs throughout the body. This process ensures that cells receive the oxygen needed to produce energy, support growth, and maintain normal physiological functions.
A normal adult hemoglobin molecule consists of two alpha globin chains and two beta globin chains working together in perfect balance. This balanced structure allows hemoglobin to efficiently pick up oxygen in the lungs and release it where the body needs it most.
When hemoglobin formation is normal:
- Oxygen delivery remains efficient.
- Red blood cells survive approximately 120 days.
- Tissues receive adequate oxygen.
- Organs function properly.
- Energy production remains stable.
- Bone marrow produces healthy red blood cells.
Healthy hemoglobin synthesis is therefore critical for maintaining normal blood health and overall well-being.
However, Alpha and beta globin synthesis defects interfere with the proper assembly of hemoglobin molecules. When either alpha or beta globin production is reduced due to inherited genetic mutations, excess unmatched globin chains accumulate inside developing red blood cells. These unstable chains can damage cell membranes and trigger premature cell destruction.
As a result:
- Red blood cells become fragile.
- Oxygen delivery declines.
- Bone marrow compensates by producing more cells.
- Chronic anemia develops.
- Fatigue and weakness become common.
- The spleen may enlarge due to increased red blood cell destruction.
Over time, the body attempts to compensate for low oxygen levels by increasing red blood cell production. Unfortunately, many of these newly formed cells are abnormal and are destroyed before they can function effectively. This ongoing cycle contributes to the chronic complications commonly associated with thalassemia.
This chain of events explains many symptoms experienced by thalassemia patients, including persistent fatigue, shortness of breath, pale skin, growth delays, and reduced exercise tolerance. The severity of symptoms often depends on the degree of Alpha and Beta Globin Chain Imbalance and the specific genetic mutations involved.
For a deeper understanding of hemoglobin disorders and their impact on blood health, read our guide on Hemoglobin Synthesis Disorders.
How Alpha and Beta Globin Chain Imbalance Occurs
Alpha and Beta Globin Chain Imbalance develops when genetic mutations affect the body’s ability to produce normal amounts of alpha or beta globin chains. Since healthy hemoglobin requires a balanced ratio of these chains, any disruption can interfere with red blood cell development and function.
The imbalance can occur in two primary forms: alpha thalassemia and beta thalassemia. Although both conditions involve abnormal globin production, the mechanisms and clinical effects differ depending on which chain is affected.
Alpha Thalassemia
Alpha thalassemia develops when one or more alpha globin genes are deleted, damaged, or mutated. Since alpha globin genes are responsible for producing alpha chains, any reduction in their activity results in decreased alpha chain production.
Because fewer alpha chains are produced:
- Beta chains accumulate.
- Hemoglobin structure becomes abnormal.
- Oxygen transport efficiency decreases.
- Red blood cells become less stable.
- Anemia may develop.
The severity of alpha thalassemia depends on the number of affected alpha globin genes. Humans normally inherit four alpha globin genes, two from each parent. The more genes affected, the greater the degree of Globin chain imbalance in thalassemia.
One Gene Missing
When only one alpha globin gene is affected, individuals are typically silent carriers.
Characteristics include:
- Usually no symptoms
- Normal life expectancy
- Often discovered only through genetic testing
Two Genes Missing
Loss of two alpha globin genes usually results in alpha thalassemia trait.
Possible effects include:
- Mild anemia
- Slightly reduced hemoglobin levels
- Small red blood cells (microcytosis)
- Few or no noticeable symptoms
Many individuals remain unaware of their condition unless they undergo specialized blood testing.
Three Genes Missing
When three alpha globin genes are missing or defective, the condition is known as Hemoglobin H disease.
Common complications include:
- Moderate to severe anemia
- Enlarged spleen
- Fatigue and weakness
- Growth delays in children
- Increased risk of hemolysis
At this stage, significant Thalassemia globin chain abnormalities interfere with normal oxygen transport and may require ongoing medical monitoring.
Four Genes Missing
Loss of all four alpha globin genes results in the most severe form of alpha thalassemia.
This condition causes:
- Severe fetal anemia
- Heart failure before birth
- Generalized swelling (hydrops fetalis)
- High risk of fetal or newborn death without intervention
Because no functional alpha chains are produced, normal hemoglobin cannot form, making this form of Alpha and Beta Globin Chain Imbalance life-threatening. Advances in prenatal diagnosis and specialized fetal treatment have improved outcomes in some cases, but the condition remains extremely serious.
For additional information about alpha thalassemia and inherited carrier conditions, visit Alpha Thalassemia Carrier Pregnancy Guide and Types of Thalassemia: Alpha vs Beta.
Beta Thalassemia
Beta thalassemia results from mutations affecting beta globin production.
When beta chains decrease:
- Excess alpha chains accumulate.
- Alpha chains damage developing red blood cells.
- Severe ineffective erythropoiesis occurs.
This form of Alpha and Beta Globin Chain Imbalance is particularly destructive because excess alpha chains are highly unstable.
Learn more about the underlying causes:
https://thalassemiaawarenet.com/beta-thalassemia-causes-complete-guide/
The Genetics Behind Globin Chain Imbalance
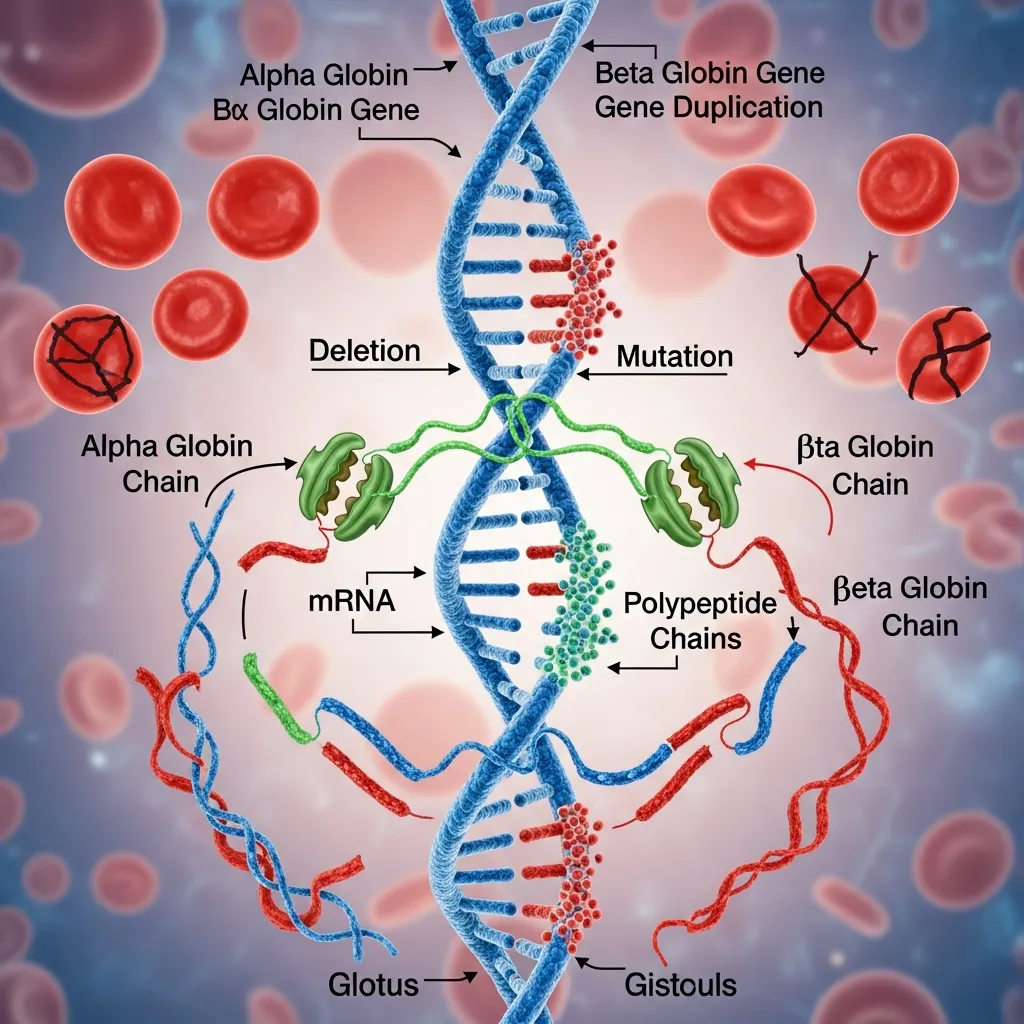 The inheritance pattern of thalassemia follows an autosomal recessive model, which means that a child must inherit altered genes from both parents to develop a clinically significant form of the disease. If only one defective gene is inherited, the individual usually becomes a carrier without severe symptoms, although mild anemia or microcytosis may still be present in some cases.
The inheritance pattern of thalassemia follows an autosomal recessive model, which means that a child must inherit altered genes from both parents to develop a clinically significant form of the disease. If only one defective gene is inherited, the individual usually becomes a carrier without severe symptoms, although mild anemia or microcytosis may still be present in some cases.
In this genetic model:
- A child inherits one gene from each parent.
- Two abnormal genes usually produce significant disease.
- One abnormal gene often results in carrier status.
This pattern is a key reason why Alpha and Beta Globin Chain Imbalance is frequently seen in families with a history of thalassemia or hemoglobin disorders. When both parents carry mutated genes, there is a higher probability that their children will inherit more severe forms of the condition.
Genetic mutations can reduce globin production in several ways:
- Reduced gene expression
- Decreased protein production
- Abnormal protein stability
These disruptions lead to varying levels of Thalassemia globin chain abnormalities, depending on how severely the gene function is affected. Even small changes in gene expression can significantly impact hemoglobin production and red blood cell health.
Common mutation types include:
- Gene deletions
- Point mutations
- Promoter mutations
- Splicing mutations
Each type of mutation affects globin production differently, which explains why thalassemia presents in a wide spectrum—from silent carriers to severe transfusion-dependent anemia. For a deeper understanding of mutation mechanisms, visit Thalassemia Genetic Mutation Guide.
Pathophysiology of Globin Chain Imbalance
The pathophysiology of Alpha and Beta Globin Chain Imbalance is primarily driven by the unequal production of globin chains, which disrupts hemoglobin assembly and leads to ineffective red blood cell formation.
Excess Alpha Chains
In beta thalassemia, beta globin production is reduced or absent, while alpha globin production continues at a normal or near-normal rate. This imbalance results in excess free alpha chains inside developing red blood cells.
These excess alpha chains are highly unstable and tend to form toxic aggregates. These aggregates have several harmful effects:
- Damage red blood cell membranes
- Trigger oxidative stress inside cells
- Disrupt normal hemoglobin assembly
- Cause premature destruction of red blood cell precursors
As a result, many developing red blood cells die within the bone marrow before they can mature and enter circulation. This condition is a major contributor to anemia severity in beta thalassemia patients.
Over time, the persistent destruction of red blood cell precursors leads to chronic anemia and stimulates the bone marrow to increase red blood cell production, further worsening the imbalance.
Excess Beta Chains
In alpha thalassemia, the opposite situation occurs. Alpha globin production is reduced, leading to an excess of beta chains. These beta chains can form abnormal tetramers such as hemoglobin H (HbH).
While these abnormal structures can still carry oxygen, they are not efficient and tend to destabilize red blood cells. However, compared to excess alpha chains, excess beta chains are generally less toxic.
Despite this difference, alpha thalassemia can still cause significant health problems depending on how many alpha genes are affected. This imbalance contributes to reduced oxygen delivery, anemia, and complications in moderate to severe cases.
This difference in toxicity explains why beta thalassemia often presents with more severe clinical symptoms than many forms of alpha thalassemia.
Ineffective Erythropoiesis and Bone Marrow Expansion
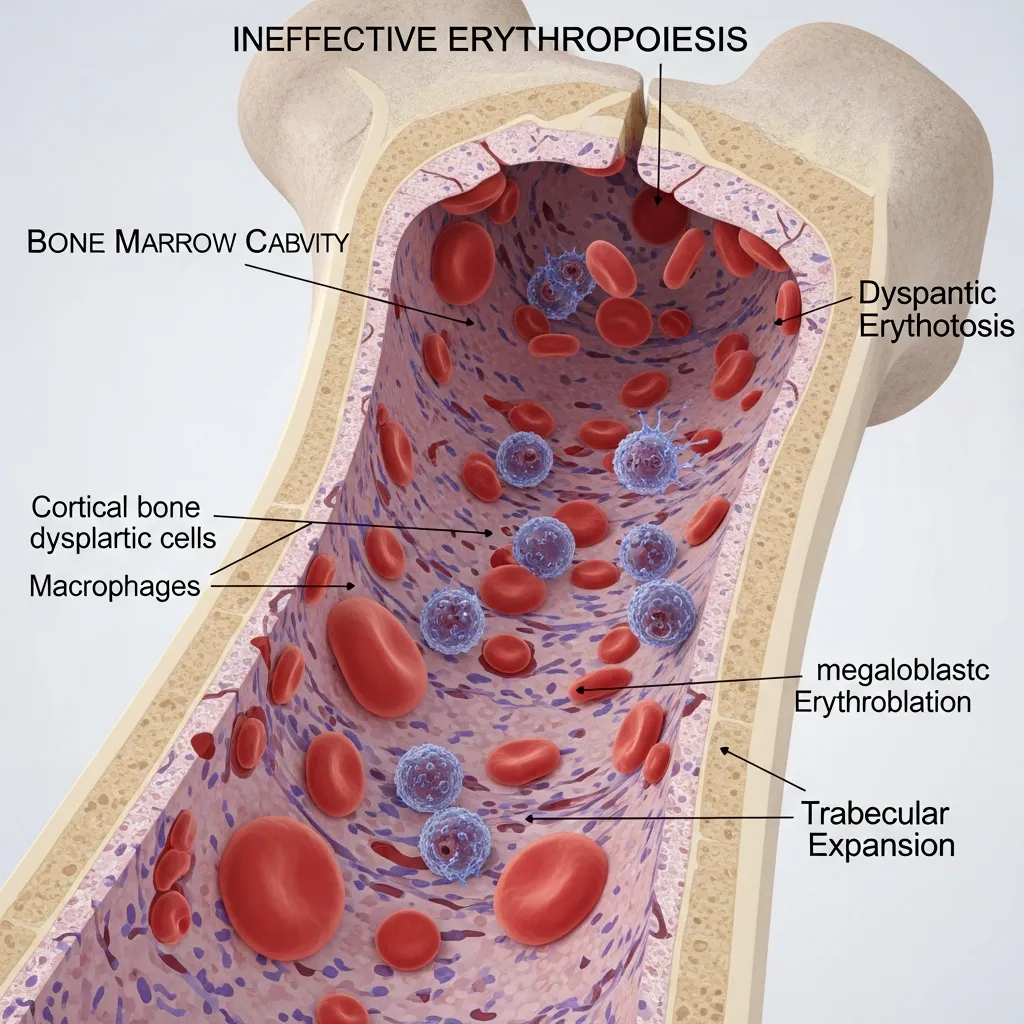 One of the most important consequences of Alpha and Beta Globin Chain Imbalance is ineffective erythropoiesis, a condition where red blood cell production in the bone marrow is severely disrupted.
One of the most important consequences of Alpha and Beta Globin Chain Imbalance is ineffective erythropoiesis, a condition where red blood cell production in the bone marrow is severely disrupted.
Normally, the bone marrow produces healthy red blood cells that enter circulation and survive for about 120 days. However, in thalassemia:
- The bone marrow attempts to produce red blood cells continuously.
- A large proportion of developing cells are destroyed prematurely.
- Only a small number of mature cells reach circulation.
This inefficiency forces the body to compensate by dramatically increasing bone marrow activity. Over time, this excessive stimulation leads to structural changes in bones and other complications.
Common consequences include:
- Bone deformities, especially in the face and skull
- Enlarged facial bones
- Osteoporosis and reduced bone density
- Growth abnormalities in children
- Expansion of bone marrow cavities
These skeletal changes are particularly noticeable in untreated or severe forms of thalassemia. Ineffective erythropoiesis also increases iron absorption from the gut, which may lead to iron overload even in patients who are not receiving frequent blood transfusions.
For more detailed insight into this mechanism, read Ineffective Erythropoiesis in Thalassemia.
Symptoms of Alpha and Beta Globin Chain Imbalance
The symptoms of Alpha and Beta Globin Chain Imbalance vary widely depending on the severity of the genetic defect and the type of thalassemia present. Some individuals may remain asymptomatic carriers, while others may develop severe, life-threatening anemia.
Common Symptoms
Chronic Fatigue
Reduced oxygen delivery leads to persistent tiredness, weakness, and lack of energy.
Pale Skin
Anemia often causes noticeable paleness due to reduced hemoglobin levels.
Shortness of Breath
Low oxygen supply makes physical activity difficult and increases breathing effort.
Dizziness and Headaches
Reduced oxygen to the brain can cause frequent dizziness and cognitive slowing.
Exercise Intolerance
Patients may feel exhausted even after mild physical activity.
Rapid Heartbeat (Tachycardia)
The heart works harder to compensate for reduced oxygen transport.
Severe Symptoms
In more advanced cases of Globin chain imbalance in thalassemia, symptoms become more serious and may include:
- Enlarged spleen (splenomegaly)
- Enlarged liver (hepatomegaly)
- Delayed growth and puberty in children
- Bone deformities and facial changes
- Severe chronic anemia requiring transfusions
These symptoms reflect the long-term effects of ineffective erythropoiesis and chronic oxygen deprivation.
How Doctors Diagnose Alpha and Beta Globin Chain Imbalance
Accurate diagnosis of Alpha and Beta Globin Chain Imbalance requires a combination of blood tests, microscopic analysis, and genetic studies.
Complete Blood Count (CBC)
A CBC is usually the first diagnostic test performed. It evaluates:
- Hemoglobin concentration
- Red blood cell count
- Mean corpuscular volume (MCV)
Patients with thalassemia often show microcytic (small-sized) and hypochromic (pale) red blood cells, which raises suspicion for a globin disorder.
Peripheral Blood Smear
A blood smear allows direct visualization of red blood cells under a microscope. Common findings include:
- Target cells
- Microcytosis
- Hypochromia
- Abnormally shaped red blood cells
These findings help support a diagnosis of thalassemia.
Hemoglobin Electrophoresis
This test separates different types of hemoglobin and identifies abnormal patterns. It is essential for distinguishing between alpha and beta thalassemia and other hemoglobin disorders.
Genetic Testing
Genetic testing provides the most definitive diagnosis. It can:
- Identify specific gene mutations
- Confirm carrier status
- Predict disease severity
- Guide family planning decisions
Hemoglobin Panel Testing
A hemoglobin panel provides a detailed analysis of globin chain production and hemoglobin variants. It is especially useful for treatment planning and long-term monitoring.
For more information, visit Hemoglobin Panel Test Guide.
Conclusion
Alpha and Beta Globin Chain Imbalance plays a central role in the development and progression of thalassemia and other hemoglobin disorders. When the production of alpha and beta globin chains becomes unbalanced, hemoglobin structure is disrupted, leading to ineffective red blood cell formation, chronic anemia, and a wide spectrum of clinical complications.
From genetic mutations to bone marrow dysfunction, the entire disease process is driven by this fundamental imbalance. Understanding Globin chain imbalance in thalassemia helps explain why patients experience symptoms such as fatigue, growth delays, organ enlargement, and bone changes.
Frequently Asked Questions (FAQs)
1. What is Alpha and Beta Globin Chain Imbalance?
It is a condition where alpha and beta globin chains are produced in unequal amounts, leading to abnormal hemoglobin formation and thalassemia.
2. What causes globin chain imbalance?
It is mainly caused by genetic mutations or deletions in alpha or beta globin genes inherited from parents.
3. How does globin chain imbalance affect red blood cells?
It leads to unstable hemoglobin, premature destruction of red blood cells, and reduced oxygen delivery in the body.
4. Is Alpha and Beta Globin Chain Imbalance genetic?
Yes, it is an inherited autosomal recessive genetic disorder passed from parents to children.
5. What are the main symptoms of globin chain imbalance?
Common symptoms include fatigue, pale skin, shortness of breath, dizziness, and, in severe cases, organ enlargement and bone changes.
6. How is thalassemia related to globin chain imbalance?
Thalassemia is directly caused by Globin chain imbalance, in which one type of globin chain is underproduced.
7. How is the condition diagnosed?
It is diagnosed using CBC tests, blood smears, hemoglobin electrophoresis, and genetic testing.
8. Can globin chain imbalance be treated?
While it cannot always be cured, treatments like blood transfusions, iron chelation, and gene therapy can help manage it.
9. What is the difference between alpha and beta thalassemia?
Alpha thalassemia involves reduced alpha chain production, while beta thalassemia involves reduced beta chain production.
10. Can lifestyle changes help manage this condition?
Yes, proper nutrition, regular monitoring, and avoiding iron overload can help improve overall health in patients.