


Hemoglobin E Disease is an inherited blood disorder that affects red blood cells and hemoglobin production. It may cause mild to moderate anemia, fatigue, and weakness. Early diagnosis, regular monitoring, and proper treatment help manage symptoms and improve overall quality of life.
Fatigue, weakness, and an unexplained pale complexion can sometimes be dismissed as simple exhaustion or poor diet. However, these symptoms can occasionally point to a much deeper genetic blood condition. Millions of people worldwide carry genetic variations that alter how their red blood cells function, and one of the most significant yet frequently misunderstood of these is hemoglobin E disease.
Hemoglobin is the vital iron-rich protein inside your red blood cells responsible for transporting oxygen from your lungs to every tissue in your body. When a person inherits specific genetic mutations, the structure of this protein changes. Hemoglobin E disease occurs when this mutated protein replaces normal hemoglobin, causing the red blood cells to become fragile, smaller than usual, and less effective at carrying oxygen. This leads to varying degrees of anemia and a host of other potential medical complications.
While the condition is found globally, hemoglobin E disease has a particularly high prevalence in Southeast Asia, where carrier rates in some populations can reach up to 60 percent. As global migration increases, healthcare providers worldwide are seeing more cases of this disorder. Recognizing the symptoms and understanding the genetic factors behind it is crucial for anyone affected by the condition, as well as for families planning for the future.
This comprehensive guide covers everything you need to know about hemoglobin E disease. We will explore the genetic causes, the different types of the condition, its symptoms, how doctors diagnose it, and the most effective management strategies available to help patients live healthy, active lives.
The Genetics of Hemoglobin E Disease
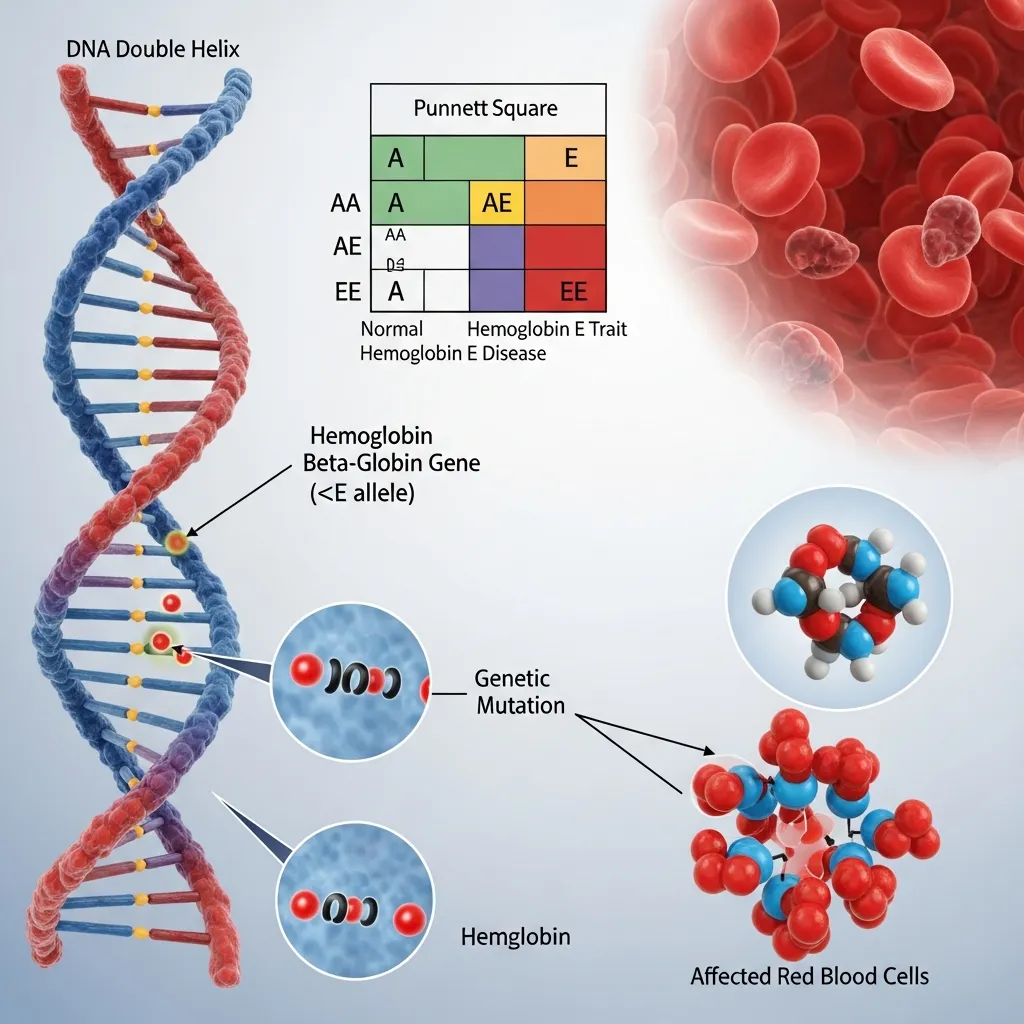 To fully understand hemoglobin E disease, we have to look closely at the body’s genetic instruction manual. Genes dictate how every protein in our body is built. When there is a spelling error in that manual, the resulting protein behaves differently.
To fully understand hemoglobin E disease, we have to look closely at the body’s genetic instruction manual. Genes dictate how every protein in our body is built. When there is a spelling error in that manual, the resulting protein behaves differently.
Hemoglobin E Gene Mutation (HBB:c.79G>A)
Normal adult hemoglobin is made up of two alpha-globin and two beta-globin protein chains. The instructions for making beta-globin are found in the HBB gene. Hemoglobin E disease is caused by a specific mutation in this gene, technically known as HBB:c.79G>A. This single genetic typo causes the substitution of the amino acid lysine for glutamic acid in the beta-globin chain.
Because of this slight structural change, the body produces hemoglobin E instead of normal hemoglobin A. Furthermore, this mutation also affects how the gene is processed, leading to a mild reduction in the overall amount of beta-globin produced.
Inheritance Patterns
Hemoglobin E disease follows an autosomal recessive inheritance pattern. This means a person must inherit two mutated genes—one from each parent—to have the full disease.
If a child inherits one normal hemoglobin A gene and one hemoglobin E gene, they have the heterozygous state, commonly known as hemoglobin E trait. These individuals are carriers. If a child inherits a hemoglobin E gene from both parents, they have the homozygous state, which is classic hemoglobin E disease.
Things become more complicated when a person inherits one hemoglobin E gene from one parent and a different beta-globin mutation (like beta-thalassemia) from the other. This creates a compound heterozygous state, leading to a much more severe condition known as hemoglobin E/beta-thalassemia. For more details on these inheritance patterns, the Centers for Disease Control and Prevention (CDC) provides excellent resources on hemoglobin disorders.
Genetic Counseling and Testing
Because carriers of the hemoglobin E trait often show no symptoms, they usually do not know they carry the mutation until they undergo blood testing. Genetic counseling is highly recommended for prospective parents, especially those with Southeast Asian heritage. A genetic counselor can explain the risks of passing hemoglobin E disease to children and help families navigate prenatal testing options.
Types of Hemoglobin E Conditions
The clinical impact of the hemoglobin E mutation varies wildly depending on which combination of genes a person inherits. Medical professionals generally divide the condition into three distinct categories.
Hemoglobin E Trait (Hb AE)
Individuals with hemoglobin E trait carry one normal gene and one mutated gene. They produce both normal hemoglobin A and hemoglobin E. The condition is almost always benign. Most people with hemoglobin E trait live completely normal lives and require no medical treatment. Their red blood cells might be slightly smaller than average, but they do not experience significant anemia.
Hemoglobin E Disease (Hb EE)
When a person inherits the mutated gene from both parents, they develop classic hemoglobin E disease. These individuals produce no normal hemoglobin A; their red blood cells contain almost entirely hemoglobin E. Because hemoglobin E is slightly unstable and produced in lower quantities, these patients typically experience mild anemia. They may also have a slightly enlarged spleen. However, hemoglobin E disease is generally considered a mild disorder, and many patients require little to no medical intervention.
Hemoglobin E/Beta-Thalassemia (Hb E/β-Thal)
This is the most clinically significant and severe form of the condition. It occurs when a person inherits a hemoglobin E gene from one parent and a beta-thalassemia gene from the other. You can learn more about how beta-thalassemia affects the body by reading our guide on Understanding Thalassemia: Causes, Types, Symptoms, and Treatment.
Hemoglobin E/beta-thalassemia is a highly variable disease. It manifests across a wide spectrum of severity:
Mild to Moderate Forms
Some patients with hemoglobin E/beta-thalassemia experience moderate anemia. They might feel fatigued and have an enlarged spleen, but they do not require regular blood transfusions to survive. They are often monitored closely by a hematologist and only receive transfusions during times of extreme stress, such as a severe infection or pregnancy.
Severe Forms and Clinical Implications
On the other end of the spectrum, some patients develop severe, life-threatening anemia that closely resembles beta-thalassemia major. These individuals require lifelong, regular blood transfusions starting from early childhood to maintain their hemoglobin levels and support normal physical growth.
Symptoms and Clinical Manifestations
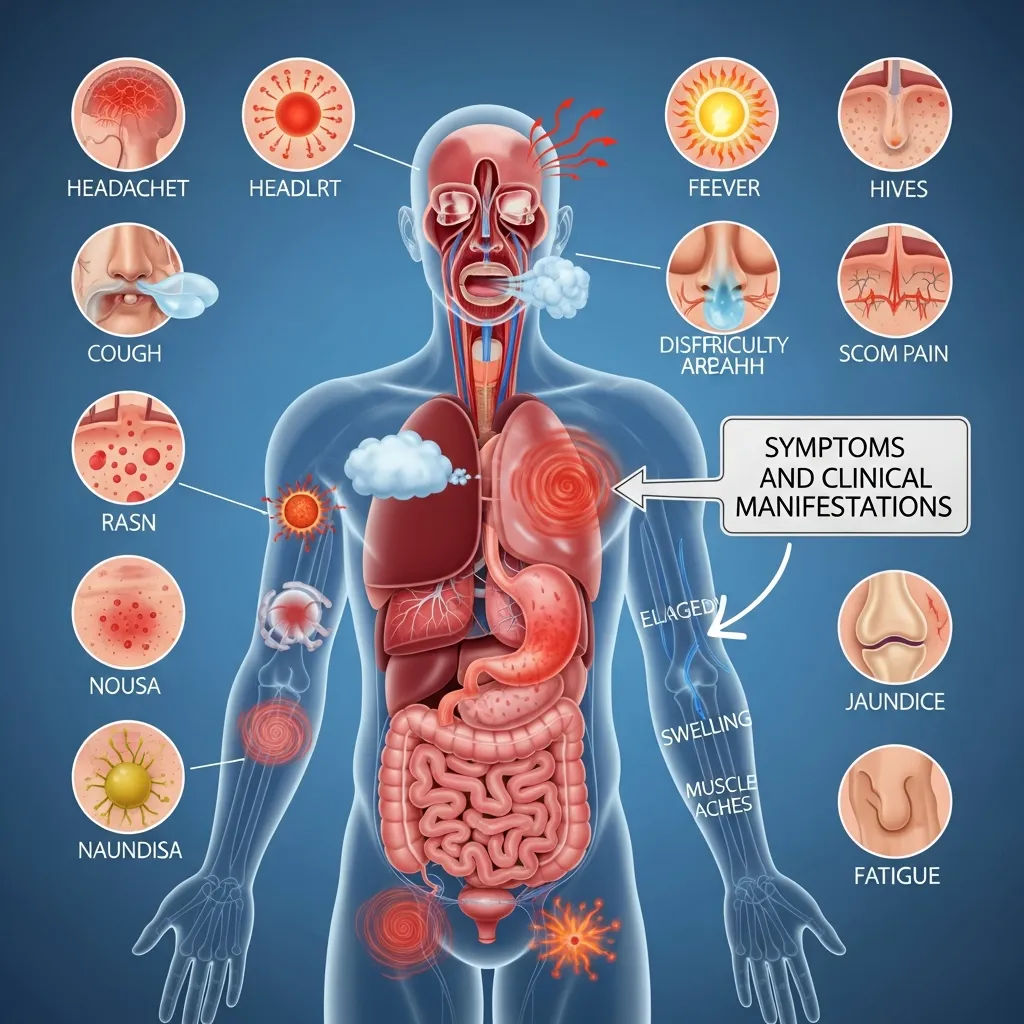 The signs of a hemoglobin disorder depend entirely on the specific genetic type the patient has inherited.
The signs of a hemoglobin disorder depend entirely on the specific genetic type the patient has inherited.
Hemoglobin E Trait: Often Asymptomatic
As mentioned earlier, the trait form is asymptomatic. Routine blood work might show slightly smaller red blood cells, which can sometimes be misdiagnosed as mild iron deficiency. Otherwise, carriers experience no physical symptoms.
Hemoglobin E Disease: Mild Anemia and Splenomegaly
For those with homozygous hemoglobin E disease, symptoms are usually mild. The most common manifestation is mild microcytic anemia, meaning the red blood cells are small and lack sufficient hemoglobin. Some patients may also develop mild splenomegaly (an enlarged spleen) because the spleen has to work slightly harder to filter out the abnormally shaped red blood cells. Fatigue might occur occasionally, but it rarely interferes with daily life.
Hemoglobin E/Beta-Thalassemia: A Spectrum of Severity
Patients with the compound hemoglobin E/beta-thalassemia face a much wider and more challenging array of symptoms.
Anemia (Microcytic, Hypochromic)
The most prominent symptom is chronic anemia. The body fails to produce enough healthy red blood cells, leading to a profound lack of oxygen in the tissues. Patients experience severe fatigue, weakness, pale skin, and shortness of breath. To understand how doctors categorize these different types of anemia, review our guide on Understanding Anemia Classification.
Splenomegaly and Hepatomegaly
Because the red blood cells are defective, the spleen works overtime to clear them from the bloodstream. This causes the spleen to expand massively (splenomegaly). The liver may also become enlarged (hepatomegaly) as it tries to compensate for the bone marrow’s inability to produce healthy blood cells.
Jaundice and Gallstones
The rapid destruction of red blood cells releases high levels of bilirubin into the blood. This excess bilirubin causes jaundice, turning the skin and the whites of the eyes yellow. High bilirubin levels also significantly increase the risk of developing painful gallstones.
Bone Changes and Growth Retardation
In severe cases, the bone marrow attempts to combat the extreme anemia by expanding rapidly to produce more blood cells. This expansion causes the bones to become thin, brittle, and deformed. Children with severe hemoglobin E/beta-thalassemia may develop prominent facial bones and experience severely delayed physical growth and puberty.
Iron Overload and its Complications
Patients who require frequent blood transfusions face a secondary, highly dangerous problem: iron overload. The human body has no effective way to excrete the excess iron introduced by transfused blood. This iron accumulates in the heart, liver, and endocrine glands, eventually leading to heart failure, liver cirrhosis, and diabetes if left untreated.
Diagnosis of Hemoglobin E Disease
Accurate diagnosis is critical. Because mild forms of hemoglobin E disease look very similar to basic iron deficiency anemia under a microscope, specialized testing is required to pinpoint the exact genetic cause. If you suspect a blood disorder, it is vital to consult a specialist. Our guide on finding the right anemia treatment doctor can help you take the first steps.
Complete Blood Count (CBC) and Red Blood Cell Indices
The diagnostic process usually begins with a simple Complete Blood Count (CBC). This test measures the number of red blood cells, the amount of hemoglobin, and the size of the cells (Mean Corpuscular Volume, or MCV). In hemoglobin E disease, the MCV is almost always lower than normal.
Hemoglobin Electrophoresis and HPLC
To confirm the diagnosis, doctors must look at the specific types of hemoglobin present in the blood. Hemoglobin electrophoresis and High-Performance Liquid Chromatography (HPLC) are laboratory tests that separate the different hemoglobin proteins. These tests easily identify the presence of hemoglobin E and determine whether normal hemoglobin A is also present.
Genetic Testing and DNA Analysis
For a definitive diagnosis, especially when determining the exact mutations in a case of hemoglobin E/beta-thalassemia, DNA analysis is used. Genetic testing maps out the HBB gene, providing exact details about the mutations. This is incredibly helpful for predicting disease severity and guiding family planning.
Differential Diagnosis
Doctors must carefully distinguish hemoglobin E disease from other conditions. Iron deficiency anemia is the most common misdiagnosis. Prescribing iron supplements to a person with hemoglobin E disease can be harmful, as they do not need extra iron and may already be prone to iron accumulation. The diagnostic tests mentioned above easily separate these two conditions.
Management and Treatment Strategies
 Treatment for hemoglobin E disease is highly individualized. Doctors design care plans based entirely on how severely the anemia affects the patient.
Treatment for hemoglobin E disease is highly individualized. Doctors design care plans based entirely on how severely the anemia affects the patient.
Hemoglobin E Trait and Asymptomatic Hemoglobin E Disease
Patients with the trait or the mild homozygous disease generally need no medical treatment. Doctors simply recommend routine monitoring during annual checkups. Folic acid supplements may be suggested to support red blood cell production, but otherwise, patients are encouraged to live normal lives.
Management of Hemoglobin E/Beta-Thalassemia
For patients on the severe end of the spectrum, medical care is intensive and lifelong.
Regular Blood Transfusions
Those with severe anemia require regular blood transfusions, often every three to four weeks. These transfusions suppress the body’s ineffective blood production, relieve the anemia, prevent bone deformities, and allow children to grow normally.
Iron Chelation Therapy
To combat the fatal iron overload caused by regular transfusions, patients must take chelation medications. These drugs bind to the excess iron in the bloodstream, allowing the body to safely excrete it through urine or stool. Strict adherence to chelation therapy is the most critical factor in extending the lifespan of transfusion-dependent patients.
Splenectomy
If the spleen becomes too large, it traps and destroys healthy transfused blood cells, making transfusions less effective. In these cases, doctors may recommend a splenectomy (surgical removal of the spleen). While this reduces transfusion requirements, it significantly increases the patient’s risk of severe infections.
Folic Acid Supplementation
Because the bone marrow is working so hard to produce cells, it rapidly consumes folic acid. Daily folic acid supplements help support this intense cellular activity.
Bone Marrow Transplantation
Currently, a bone marrow or stem cell transplant is the only established cure for severe hemoglobin E/beta-thalassemia. By replacing the patient’s defective bone marrow with healthy stem cells from a matched donor, the patient can begin producing normal hemoglobin. The procedure carries high risks and requires a perfectly matched donor, usually a sibling.
Emerging Therapies and Clinical Trials
Medical science is advancing rapidly. New medications designed to help red blood cells mature properly are currently entering the market, potentially reducing the need for transfusions. The National Institutes of Health (NIH) provides a database of ongoing clinical trials where patients can access these cutting-edge therapies.
Living with Hemoglobin E Disease: Quality of Life and Prognosis
A chronic diagnosis changes how a person approaches daily life, but it does not have to stop them from achieving their goals.
Lifestyle Adjustments and Dietary Considerations
Nutrition plays a supportive role in disease management. Patients with severe forms must avoid iron-rich foods like red meat and fortified cereals to prevent worsening their iron overload. Instead, they should focus on a balanced diet rich in calcium and vitamin D to protect their bones, which are often weakened by the disease.
Importance of Regular Medical Follow-ups
Consistency is key. Patients must attend all scheduled medical appointments to monitor their hemoglobin levels, track iron accumulation through MRI scans, and check the health of their heart and liver. Catching complications early drastically improves long-term outcomes.
Psychological and Social Support
Managing a lifelong illness takes an emotional toll. The anxiety of frequent hospital visits and the physical fatigue can lead to depression. Connecting with support groups, talking to mental health professionals, and maintaining open communication with family members helps patients navigate the heavy mental burden of hemoglobin E disease.
Prevention and Public Health Initiatives
Because this is a genetic condition, public health strategies focus heavily on education and informed family planning.
Carrier Screening Programs
In regions where the hemoglobin E mutation is common, routine carrier screening is a vital public health tool. Simple blood tests can identify carriers before they start a family, providing them with crucial information about their reproductive risks.
Prenatal Diagnosis and Genetic Counseling
When two carriers are expecting a child, prenatal diagnosis techniques like chorionic villus sampling (CVS) or amniocentesis can determine if the fetus has inherited the severe form of the disease. Genetic counselors help parents understand these results and prepare for the medical journey ahead.
Education and Awareness Campaigns
Raising awareness about hemoglobin E disease helps reduce the stigma associated with genetic disorders. Education encourages more people to get tested and highlights the constant need for voluntary blood donations, which keep severe patients alive.
Research and Future Directions
The future of treatment for hemoglobin disorders is incredibly bright. Scientists are moving away from simply managing symptoms and are working directly to fix the genetic code.
Advances in Gene Therapy
Gene therapy is fundamentally changing the landscape of blood disorders. By collecting a patient’s own stem cells, correcting the faulty HBB gene in a laboratory, and reinfusing the cells back into the body, scientists are offering functional cures. You can read more about this revolutionary science in our article on Gene Therapy for Thalassemia.
New Drug Development
Pharmaceutical companies are developing novel drugs that target the specific biological pathways causing ineffective red blood cell production. These drugs aim to boost the body’s natural ability to create healthy cells, thereby reducing or entirely eliminating the need for lifelong blood transfusions.
Personalized Medicine
As genetic profiling becomes faster and cheaper, doctors will soon be able to tailor treatment plans to the exact molecular signature of a patient’s specific hemoglobin E disease. This personalized medicine approach ensures that patients receive the most effective therapies with the fewest side effects.
Navigating the Path Forward
 Hemoglobin E disease represents a complex intersection of genetics and human health. Ranging from a completely harmless trait to a severe, life-altering condition, its impact on the body is profound. The sheer variability of the disease highlights exactly why accurate diagnostic testing and specialized medical care are so crucial.
Hemoglobin E disease represents a complex intersection of genetics and human health. Ranging from a completely harmless trait to a severe, life-altering condition, its impact on the body is profound. The sheer variability of the disease highlights exactly why accurate diagnostic testing and specialized medical care are so crucial.
By understanding the genetic roots, recognizing the symptoms, and strictly adhering to modern medical management like blood transfusions and chelation therapy, patients can effectively protect their vital organs and sustain their energy. Advancements in gene therapy and personalized medicine mean that the next generation of patients will have even better tools at their disposal, transforming what was once a devastating diagnosis into a highly manageable, and potentially curable, condition. Early detection and proactive care remain your strongest defense, ensuring a healthier, more vibrant life despite the challenges of a chronic blood disorder.