


This comprehensive pillar content explores Thalassemia in depth, covering its genetic basis, various types like alpha and beta thalassemia (including beta thalassemia trait and pathophysiology), symptoms, diagnosis, and current treatment approaches such as thalassemia minor treatment. It also details the importance of a proper thalassemia diet and nutrition, and compares thalassemia to different kinds of blood diseases.
Thalassemia is an inherited blood disorder that affects how the body produces hemoglobin, the vital protein in red blood cells responsible for transporting oxygen throughout the body. Hemoglobin is essential for delivering oxygen to organs, muscles, and tissues. When the body cannot produce enough healthy hemoglobin, red blood cells become fragile and are destroyed faster than normal. This process leads to chronic anemia, fatigue, weakness, and a variety of other health complications that can range from mild to life-threatening.
Unlike temporary forms of anemia caused by poor nutrition or illness, thalassemia is a lifelong genetic condition passed down from parents to children through inherited gene mutations. The severity of the disorder depends on how many genes are affected and which type of hemoglobin production is impaired. Some individuals may only carry the trait without noticeable symptoms, while others may experience severe anemia requiring regular blood transfusions and ongoing medical care from early childhood.
Globally, thalassemia is considered one of the most common inherited blood disorders. It is especially prevalent among populations from the Mediterranean region, the Middle East, South Asia, Southeast Asia, and parts of Africa. As migration and multicultural populations continue to grow, thalassemia is increasingly recognized as a worldwide public health concern. Millions of people are estimated to carry thalassemia genes, often without realizing it until routine blood testing, pregnancy screening, or the birth of a child with a severe form of the disease reveals the condition.
The disorder is broadly categorized into alpha thalassemia and beta thalassemia, depending on which part of the hemoglobin molecule is affected. Each category contains several subtypes ranging from mild carrier states to severe transfusion-dependent conditions. For example, individuals with thalassemia trait may live normal lives with minimal symptoms, while patients with thalassemia major often require lifelong treatment and specialized medical monitoring.
Symptoms of thalassemia can vary significantly between individuals. Mild forms may only cause slight fatigue or pale skin, whereas severe forms can lead to growth delays, bone deformities, enlarged organs, jaundice, and complications affecting the heart, liver, and endocrine system. In children, untreated severe thalassemia can interfere with normal development and overall quality of life.
Modern medical advancements have dramatically improved survival rates and quality of life for people living with thalassemia. Treatment options may include blood transfusions, iron chelation therapy to prevent iron overload, folic acid supplementation, bone marrow transplantation, and emerging gene therapies. Additionally, maintaining a healthy lifestyle, following a balanced diet, and receiving consistent medical monitoring play an important role in managing symptoms and reducing complications.
Understanding thalassemia is essential not only for those diagnosed with the condition but also for prospective parents and families considering genetic counseling. Since carriers often show few or no symptoms, many people remain unaware of their genetic status until family planning becomes a concern. Early diagnosis, awareness, and education can help individuals make informed healthcare decisions and improve long-term outcomes.
As research continues to advance, scientists are exploring innovative therapies aimed at correcting the underlying genetic defects responsible for thalassemia. These breakthroughs offer growing hope for more effective treatments and potentially curative solutions in the future. With proper care, medical support, and awareness, many individuals with thalassemia are able to lead active, fulfilling, and productive lives despite the challenges of living with a chronic blood disorder.
The Genetics Behind Thalassemia
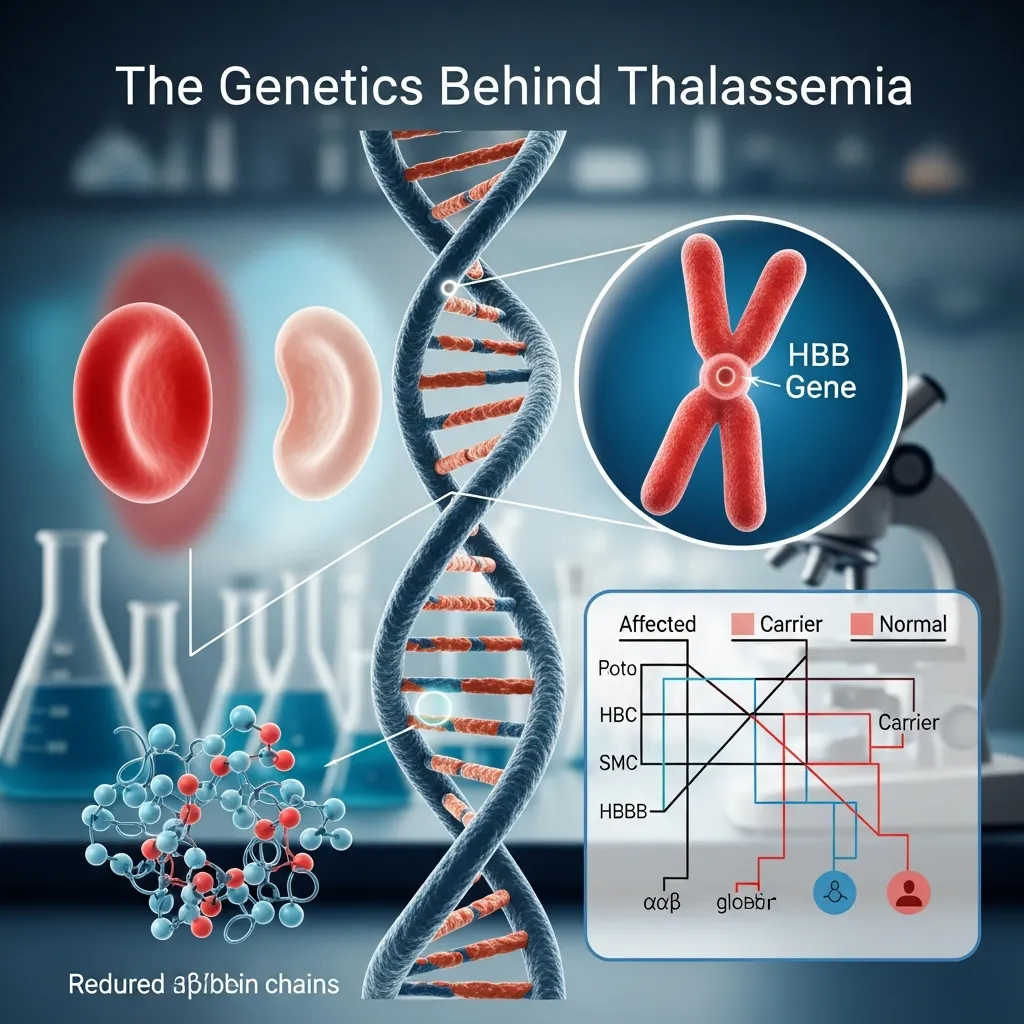 To understand thalassemia, it helps to first examine the essential role of hemoglobin in the body. Hemoglobin is the iron-rich protein found inside red blood cells that carries oxygen from the lungs to tissues and organs throughout the body. Each hemoglobin molecule is made up of two major protein chains: alpha globin and beta globin. For red blood cells to function normally, the body must produce these chains in balanced amounts.
To understand thalassemia, it helps to first examine the essential role of hemoglobin in the body. Hemoglobin is the iron-rich protein found inside red blood cells that carries oxygen from the lungs to tissues and organs throughout the body. Each hemoglobin molecule is made up of two major protein chains: alpha globin and beta globin. For red blood cells to function normally, the body must produce these chains in balanced amounts.
When genetic mutations disrupt the production of either the alpha or beta chains, hemoglobin becomes unstable and ineffective. As a result, red blood cells become fragile, misshapen, and more likely to break down prematurely. This shortened lifespan of red blood cells leads to chronic anemia, reduced oxygen delivery, and many of the complications associated with thalassemia.
Thalassemia is not caused by lifestyle choices, infections, or nutritional deficiencies. It is a hereditary condition passed from parents to children through specific gene mutations. The disease follows an autosomal recessive inheritance pattern, meaning a child usually needs to inherit defective genes from both parents to develop a severe form of the disorder.
If only one parent passes down the mutated gene, the child becomes a carrier, commonly referred to as having thalassemia trait or thalassemia minor. Carriers often live normal, healthy lives with little or no symptoms, though mild anemia may occasionally occur. However, carriers can still pass the mutation to future generations, which is why genetic counseling and screening are extremely important in high-risk populations.
The likelihood of inheritance depends heavily on the genetic status of both parents:
- If one parent is a carrier and the other is unaffected, each child has a 50% chance of inheriting the carrier trait.
If both parents carry thalassemia genes, each pregnancy carries:
- A 25% chance of having a child without thalassemia
- A 50% chance of having a carrier child
- A 25% chance of having a child with severe thalassemia
Because of these inheritance patterns, thalassemia is especially common in regions where carrier rates are high. In some Mediterranean, Middle Eastern, African, and South Asian communities, carrier frequencies can reach significant portions of the population.
Another interesting genetic aspect of thalassemia is its historical relationship with malaria. Scientists believe carriers of thalassemia traits may have had partial protection against severe malaria infections in regions where malaria was widespread. Over generations, this evolutionary advantage contributed to the higher prevalence of thalassemia genes in certain parts of the world.
Types of Thalassemia
Thalassemia is broadly categorized into two major groups depending on which hemoglobin chain is affected: alpha thalassemia and beta thalassemia. Each type can range from mild carrier states to severe, life-threatening disease.
Alpha Thalassemia
Alpha thalassemia occurs when there are defects or deletions in the genes responsible for producing alpha globin chains. Humans normally inherit four alpha-globin genes, with two genes coming from each parent. The severity of the disease depends on how many of these genes are missing or mutated.
Silent Carrier State
When only one alpha-globin gene is affected, the individual is considered a silent carrier. These individuals typically show no symptoms and often remain unaware of their genetic status throughout life. Routine blood tests may appear normal or only slightly abnormal.
Alpha Thalassemia Trait
If two alpha-globin genes are missing or defective, the person develops alpha thalassemia trait. This form usually causes mild anemia and smaller-than-normal red blood cells, a condition called microcytosis. Many individuals are mistakenly diagnosed with iron-deficiency anemia because the symptoms can appear similar.
Hemoglobin H Disease
When three alpha-globin genes are defective, the body struggles to produce enough healthy hemoglobin. This condition is called Hemoglobin H disease and often causes moderate to severe anemia. Patients may experience chronic fatigue, jaundice, enlarged spleen, bone abnormalities, and episodes of worsening anemia during infections or illness. Some individuals may require intermittent blood transfusions.
Hydrops Fetalis
The most severe form of alpha thalassemia occurs when all four alpha-globin genes are missing. This condition, known as Hydrops Fetalis or Alpha Thalassemia Major, prevents the fetus from producing functional hemoglobin. Severe oxygen deprivation develops during pregnancy, leading to life-threatening complications for both the baby and mother. Without early medical intervention, survival is extremely rare.
Beta Thalassemia
Beta thalassemia results from mutations in the HBB gene, which controls the production of beta globin chains. Unlike alpha thalassemia, humans inherit only two beta-globin genes, one from each parent.
A deeper understanding of beta thalassemia pathophysiology shows that reduced beta-globin production creates an imbalance between alpha and beta chains. Excess alpha chains accumulate inside developing red blood cells, damaging them before they fully mature. This process, known as ineffective erythropoiesis, leads to the destruction of red blood cells inside the bone marrow and contributes significantly to severe anemia.
Beta Thalassemia Minor (Trait)
Individuals with beta thalassemia trait inherit one normal beta-globin gene and one mutated gene. This condition is generally mild and may only cause slight anemia or fatigue. Many people discover their carrier status during routine blood testing or pregnancy screening.
Although symptoms are usually minimal, identifying carriers is important because two carrier parents can pass severe beta thalassemia to their children.
Beta Thalassemia Intermedia
Beta thalassemia intermedia falls between the mild and severe forms of the disease. Both beta-globin genes are affected, but the mutations still allow partial hemoglobin production. Symptoms vary widely and may include moderate anemia, delayed growth, enlarged spleen, bone deformities, gallstones, and iron overload.
Some patients can live for years without regular transfusions, while others may eventually require periodic blood transfusion therapy during stress, illness, or pregnancy.
Beta Thalassemia Major (Cooley’s Anemia)
Beta thalassemia major is the most severe and well-known form of beta thalassemia. Both beta-globin genes are severely damaged or completely inactive, leaving the body unable to produce enough healthy hemoglobin.
Babies with beta thalassemia major often appear healthy at birth because fetal hemoglobin temporarily compensates for the defective adult hemoglobin. However, symptoms usually emerge within the first two years of life as fetal hemoglobin levels decline.
Common symptoms include:
- Severe anemia
- Extreme fatigue
- Poor growth and delayed puberty
- Bone deformities, especially in the face and skull
- Enlarged liver and spleen
- Frequent infections
- Pale or yellowish skin
Without treatment, beta thalassemia major can become life-threatening. Most patients require lifelong blood transfusions combined with iron chelation therapy to remove excess iron from the body.
Rare and Uncommon Forms of Thalassemia
 In addition to the primary alpha and beta categories, several rare forms of thalassemia exist.
In addition to the primary alpha and beta categories, several rare forms of thalassemia exist.
Delta-Beta Thalassemia
Delta-beta thalassemia occurs when both delta and beta globin gene production are reduced due to genetic deletions. Symptoms are often milder than classic beta thalassemia major because the body compensates by increasing fetal hemoglobin production.
Hereditary Persistence of Fetal Hemoglobin (HPFH)
Hereditary Persistence of Fetal Hemoglobin is a rare inherited condition in which fetal hemoglobin continues to be produced into adulthood. This persistent fetal hemoglobin can partially compensate for defective adult hemoglobin and reduce the severity of symptoms in some thalassemia patients.
Combined Hemoglobin Disorders
Some individuals inherit thalassemia alongside other blood disorders, such as sickle cell disease. These combined conditions can create complex symptoms and require specialized treatment approaches tailored to the patient’s unique genetic profile.
Advances in molecular genetics and prenatal testing now allow doctors to identify many forms of thalassemia before symptoms develop. Early diagnosis enables families to better understand risks, plan future pregnancies, and begin treatment before severe complications arise.
Symptoms and Diagnosis
The symptoms of thalassemia vary greatly depending on the specific type of thalassemia and the severity of the genetic mutations involved. Some individuals may carry the gene for years without ever noticing symptoms, while others develop serious health complications during infancy or early childhood. In many cases, the severity of symptoms is directly related to how much normal hemoglobin the body is still able to produce.
People with mild forms, such as thalassemia trait or carrier states, may experience little more than slight fatigue or mild anemia. However, moderate to severe forms can significantly affect growth, organ function, physical development, and overall quality of life. Early diagnosis and consistent medical care are essential to managing complications and improving long-term outcomes.
Common Symptoms Across Types
Although symptoms differ between alpha and beta thalassemia, several signs are commonly associated with moderate and severe cases.
Chronic Anemia and Fatigue
Anemia is the hallmark symptom of thalassemia. Because the body cannot produce enough healthy red blood cells, oxygen delivery to tissues becomes inadequate. This lack of oxygen often results in:
- Persistent fatigue
- Weakness
- Reduced stamina
- Dizziness
- Shortness of breath
- Difficulty concentrating
Children with severe anemia may appear unusually tired, irritable, or less physically active than other children their age.
Pale or Yellowish Skin
A pale appearance, medically known as pallor, develops because of the reduced number of healthy red blood cells. At the same time, the rapid destruction of damaged red blood cells releases bilirubin into the bloodstream, causing jaundice. This results in yellowing of the skin and eyes, especially in more severe forms of the disease.
Bone Deformities and Skeletal Changes
The body attempts to compensate for anemia by increasing red blood cell production inside the bone marrow. This excessive bone marrow activity causes bones to expand abnormally, particularly in the face and skull.
Common skeletal changes may include:
- Prominent forehead
- Enlarged cheekbones
- Flattened nasal bridge
- Weak or brittle bones
- Delayed bone growth
Long-term bone marrow expansion may also increase the risk of osteoporosis and fractures later in life.
Enlarged Spleen and Liver
The spleen helps filter damaged red blood cells from circulation. In thalassemia, the spleen must work harder than normal, often leading to splenomegaly, or enlargement of the spleen.
An enlarged spleen may cause:
- Pain or fullness in the upper left abdomen
- Reduced appetite
- Increased destruction of blood cells
- Greater transfusion requirements
The liver may also enlarge due to increased blood cell destruction and iron accumulation.
Growth Delays and Developmental Problems
Children with untreated or poorly managed thalassemia may experience delayed physical growth and puberty. Chronic anemia reduces the amount of oxygen and nutrients available to growing tissues, which can interfere with normal development.
Possible complications include:
- Short stature
- Delayed sexual maturation
- Hormonal imbalances
- Delayed tooth development
In severe cases, nutritional deficiencies and endocrine disorders may further affect development.
Heart Complications
One of the most serious long-term complications of thalassemia is heart disease. Severe anemia forces the heart to pump harder to deliver oxygen throughout the body. In addition, repeated blood transfusions can lead to iron overload, causing iron deposits to accumulate in the heart muscle.
Potential cardiac complications include:
- Irregular heartbeat
- Enlarged heart
- Heart failure
- Cardiomyopathy
Heart-related complications remain one of the leading causes of mortality in severe transfusion-dependent thalassemia.
Iron Overload Symptoms
Patients receiving regular blood transfusions are at high risk of iron overload because the body has no natural mechanism for eliminating excess iron efficiently. Iron gradually accumulates in organs and tissues, causing progressive damage over time.
Excess iron can affect:
- The heart
- Liver
- Pancreas
- Thyroid gland
- Pituitary gland
Symptoms of iron overload may include fatigue, abdominal pain, diabetes, hormonal problems, liver disease, and skin darkening.
Increased Risk of Infections
People with thalassemia may be more vulnerable to infections, particularly if they have undergone spleen removal surgery (splenectomy). Frequent blood transfusions can also slightly increase infection risk despite modern blood screening techniques.
Vaccinations and preventive healthcare are extremely important for managing infection risk.
Symptoms in Infants and Children
 Severe forms of thalassemia, especially beta thalassemia major, often become noticeable within the first two years of life. Infants may initially appear healthy because fetal hemoglobin temporarily supports oxygen transport after birth. However, symptoms begin to develop as fetal hemoglobin levels naturally decline.
Severe forms of thalassemia, especially beta thalassemia major, often become noticeable within the first two years of life. Infants may initially appear healthy because fetal hemoglobin temporarily supports oxygen transport after birth. However, symptoms begin to develop as fetal hemoglobin levels naturally decline.
Early childhood symptoms may include:
- Poor feeding
- Slow growth
- Frequent crying or irritability
- Pale appearance
- Enlarged abdomen
- Recurrent infections
Without treatment, severe anemia can quickly become life-threatening during early childhood.
Mild and Silent Forms of Thalassemia
Not all forms of thalassemia produce noticeable symptoms. Silent carriers and individuals with thalassemia trait may never realize they carry the condition unless routine blood work reveals abnormalities.
These mild forms commonly show:
- Slight anemia
- Small red blood cells (microcytosis)
- Mild fatigue
- No major health complications
Because these signs closely resemble iron-deficiency anemia, proper testing is necessary to avoid incorrect treatment. Taking unnecessary iron supplements can be harmful in some thalassemia patients, especially if iron overload is already present.
Diagnostic Procedures
Diagnosing thalassemia usually requires a combination of laboratory testing, family history evaluation, and genetic analysis. Since symptoms overlap with many other blood disorders, specialized testing is essential for an accurate diagnosis.
Complete Blood Count (CBC)
A Complete Blood Count is typically the first test performed when anemia is suspected. This test evaluates:
- Red blood cell count
- Hemoglobin levels
- Hematocrit
- Mean corpuscular volume (MCV)
- Red blood cell size and shape
Patients with thalassemia often show low hemoglobin levels and unusually small red blood cells.
Peripheral Blood Smear
A blood smear allows doctors to examine red blood cells under a microscope. In thalassemia, red blood cells may appear:
- Smaller than normal
- Pale in color
- Irregularly shaped
- Fragile or damaged
Target cells and other abnormal red blood cell patterns are commonly seen.
Hemoglobin Electrophoresis
Hemoglobin electrophoresis is one of the most important tests for diagnosing beta thalassemia. This procedure separates the different types of hemoglobin present in the blood and measures their relative amounts.
Abnormal elevations in fetal hemoglobin (HbF) or hemoglobin A2 often suggest beta thalassemia.
High-Performance Liquid Chromatography (HPLC)
HPLC is another advanced laboratory technique used to identify and quantify abnormal hemoglobin variants. It is highly accurate and commonly used in modern diagnostic laboratories for thalassemia screening.
Iron Studies
Doctors often perform iron studies to distinguish thalassemia from iron-deficiency anemia. These tests measure:
- Serum iron
- Ferritin levels
- Total iron-binding capacity
This distinction is extremely important because iron supplementation is not always appropriate for thalassemia patients.
Genetic Testing
Genetic testing provides the most definitive diagnosis by identifying specific mutations in the alpha or beta globin genes. Molecular testing is especially valuable for:
- Confirming unclear diagnoses
- Identifying carriers
- Family planning
- Prenatal screening
Genetic counseling is strongly recommended for couples with a family history of thalassemia or known carrier status.
Prenatal and Newborn Screening
Modern prenatal testing can detect severe forms of thalassemia before birth. Common prenatal diagnostic procedures include:
Chorionic Villus Sampling (CVS)
CVS involves collecting a small sample of placental tissue during early pregnancy to analyze fetal genes.
Amniocentesis
Amniocentesis tests amniotic fluid surrounding the fetus to identify genetic abnormalities later in pregnancy.
These tests help families understand potential risks and prepare for medical care if necessary.
Many countries with high thalassemia prevalence now include newborn screening programs that allow doctors to diagnose affected infants shortly after birth. Early diagnosis enables faster treatment, prevents severe complications, and improves survival rates significantly.
Importance of Early Diagnosis
Early detection of thalassemia plays a critical role in improving long-term health outcomes. Timely diagnosis allows doctors to begin treatments such as blood transfusions, iron chelation therapy, nutritional support, and monitoring for organ complications before irreversible damage occurs.
For carriers and families, understanding genetic risks also supports informed reproductive decisions and helps reduce the incidence of severe thalassemia in future generations.
Treatment and Management
Managing thalassemia requires lifelong medical care, regular monitoring, and a personalized treatment strategy based on the type and severity of the disorder. While mild forms may need very little intervention, severe forms such as beta thalassemia major demand intensive ongoing treatment to maintain healthy red blood cell levels and prevent life-threatening complications.
Modern medical advances have significantly improved the life expectancy and quality of life for people living with thalassemia. With proper treatment, many patients are now able to attend school, work, build families, and maintain active lifestyles. However, successful management depends on consistent medical follow-up, medication adherence, nutritional support, and early detection of complications.
Blood Transfusions
For individuals with severe thalassemia, particularly Beta Thalassemia Major, regular blood transfusions are the cornerstone of treatment. These transfusions supply the body with healthy red blood cells containing normal hemoglobin, helping restore oxygen delivery throughout the body.
Transfusions help:
- Relieve severe anemia
- Reduce fatigue and weakness
- Support normal growth and development in children
- Prevent bone marrow overexpansion
- Minimize skeletal deformities
- Improve organ function and overall quality of life
Many patients require transfusions every two to five weeks depending on their hemoglobin levels and overall health status.
Long-Term Challenges of Blood Transfusions
Although blood transfusions are life-saving, they also create long-term challenges and potential complications.
Iron Overload
Each unit of transfused blood contains iron, and the body cannot naturally eliminate large excess amounts. Over time, iron accumulates in vital organs such as the heart, liver, pancreas, and endocrine glands.
Without treatment, iron overload can cause:
- Heart failure
- Liver cirrhosis
- Diabetes
- Hormonal disorders
- Delayed puberty
- Infertility
Managing iron buildup becomes one of the most important aspects of long-term thalassemia care.
Transfusion Reactions
Some patients may develop reactions during or after transfusions, including fever, allergic responses, or immune complications caused by repeated exposure to donor blood.
Risk of Infection
Modern blood screening has dramatically reduced the risk of transfusion-transmitted infections such as hepatitis and HIV. However, careful monitoring and safe transfusion practices remain essential.
Iron Chelation Therapy
 Because the human body has no efficient natural mechanism for removing excess iron, patients receiving regular blood transfusions must undergo iron chelation therapy. Chelation medications bind to excess iron and help remove it through urine or stool before it can damage vital organs.
Because the human body has no efficient natural mechanism for removing excess iron, patients receiving regular blood transfusions must undergo iron chelation therapy. Chelation medications bind to excess iron and help remove it through urine or stool before it can damage vital organs.
Iron chelation therapy is critical for preventing fatal complications associated with iron overload and has greatly improved survival rates in severe thalassemia patients.
Common Chelation Medications
Several medications are commonly used in iron chelation therapy:
Deferoxamine
Deferoxamine is one of the oldest iron chelators and is usually administered through slow injections under the skin using a pump over several hours.
Deferiprone
Deferiprone is an oral chelation medication often used when heart iron overload is a concern. It can be easier for some patients to manage compared to infusion-based therapies.
Deferasirox
Deferasirox is a once-daily oral medication widely used for long-term iron management because of its convenience and effectiveness.
Doctors regularly monitor iron levels using blood tests and imaging studies such as MRI scans to ensure chelation therapy is working properly and to adjust treatment as needed.
Monitoring Iron Levels
Patients undergoing transfusion therapy often require ongoing assessment of:
Serum ferritin levels
Liver iron concentration
Cardiac iron levels
Kidney and liver function
Careful monitoring helps prevent complications before permanent organ damage occurs.
For individuals with milder forms, such as thalassemia minor treatment, medical management is usually minimal and may not require chelation therapy at all, focusing mainly on routine monitoring and supportive care.
Bone Marrow Transplantation
Currently, a bone marrow transplant, also known as a stem cell transplant, is the only widely established cure for severe thalassemia. The procedure involves replacing the patient’s defective bone marrow with healthy stem cells from a compatible donor.
Once successful, the transplanted stem cells begin producing healthy red blood cells with normal hemoglobin.
Best Candidates for Transplantation
Bone marrow transplantation is generally most successful when:
- The patient is young
- Organ damage is still minimal
- A closely matched donor is available
- The donor is often a sibling with compatible tissue markers
Success rates are highest when the procedure is performed early in life before severe complications develop.
Risks and Challenges
Despite its curative potential, bone marrow transplantation carries serious risks, including:
- Graft-versus-host disease (GVHD)
- Severe infections
- Organ complications
- Rejection of transplanted cells
- Side effects from chemotherapy
Finding a compatible donor can also be extremely difficult for many patients.
Emerging Gene Therapy
Recent advances in gene therapy are creating new hope for individuals with severe thalassemia. Researchers are developing treatments designed to correct or replace defective globin genes directly within the patient’s own stem cells.
Some newer therapies aim to:
- Restore healthy hemoglobin production
- Reduce or eliminate transfusion dependence
- Correct the underlying genetic defect
Although still evolving and often expensive, gene therapy represents one of the most promising future directions in thalassemia treatment.
Medications and Supportive Therapies
In addition to transfusions and chelation therapy, patients may require other medications and supportive treatments to manage symptoms and complications.
Folic Acid Supplements
Folic acid helps support red blood cell production and is commonly recommended for patients with increased bone marrow activity.
Hormone Therapy
Some patients develop endocrine problems due to iron overload affecting hormone-producing glands. Hormone replacement therapy may be needed for delayed puberty, thyroid dysfunction, or fertility issues.
Antibiotics and Vaccinations
Individuals with thalassemia, especially those who have undergone spleen removal surgery, may need preventive antibiotics and routine vaccinations to reduce infection risk.
Pain Management
Bone pain, joint discomfort, and complications from enlarged organs may require supportive pain management strategies.
Surgical Management
Certain complications of thalassemia may require surgical intervention.
Splenectomy
If the spleen becomes excessively enlarged or destroys too many red blood cells, doctors may recommend spleen removal surgery, known as splenectomy.
While splenectomy can reduce transfusion needs, it also increases vulnerability to infections, making vaccinations and preventive care extremely important afterward.
Gallbladder Surgery
Chronic red blood cell destruction increases the risk of gallstones, and some patients may eventually require gallbladder removal.
The Role of Nutrition
Although nutrition cannot cure the genetic defect responsible for thalassemia, a healthy diet plays an essential role in supporting overall well-being, immune function, and organ health.
A carefully planned thalassemia diet and nutrition strategy can help manage symptoms, reduce complications, and improve energy levels.
Managing Iron Intake
Patients with iron overload are often advised to avoid excessive dietary iron intake, especially from:
- Red meat
- Iron-fortified supplements
- Iron-rich processed foods
However, dietary restrictions should always be guided by a healthcare professional because nutritional needs vary between individuals.
Importance of Calcium and Vitamin D
Bone weakness and osteoporosis are common complications in thalassemia. Calcium and vitamin D help strengthen bones and reduce fracture risk.
Good dietary sources include:
- Dairy products
- Leafy green vegetables
- Fortified foods
- Safe sunlight exposure
Folic Acid and Nutritional Support
Folic acid supports healthy red blood cell production and may help reduce fatigue and weakness in some patients.
Antioxidants and Immune Support
Fruits, vegetables, whole grains, and antioxidant-rich foods may help support immune health and reduce oxidative stress caused by chronic anemia and iron overload.
Lifestyle and Long-Term Monitoring
 Living with thalassemia requires ongoing medical monitoring throughout life. Patients often work closely with hematologists, cardiologists, endocrinologists, nutritionists, and other specialists to manage complications effectively.
Living with thalassemia requires ongoing medical monitoring throughout life. Patients often work closely with hematologists, cardiologists, endocrinologists, nutritionists, and other specialists to manage complications effectively.
Routine monitoring may include:
- Blood tests
- Heart evaluations
- Liver function tests
- Bone density scans
- Hormone assessments
- MRI imaging for iron overload
Regular exercise, adequate sleep, stress management, and emotional support also contribute significantly to long-term health and quality of life.
Psychological and Emotional Support
Chronic illnesses like thalassemia can affect emotional and mental well-being. Frequent hospital visits, lifelong treatments, and physical symptoms may contribute to anxiety, depression, or emotional stress.
Support from family, counselors, patient advocacy groups, and thalassemia communities can provide valuable emotional encouragement and practical guidance for both patients and caregivers.
With modern treatment options, early diagnosis, and comprehensive care, many individuals with thalassemia now live longer and healthier lives than ever before. Continued medical research and advancements in gene therapy continue to offer hope for even more effective and potentially curative treatments in the future.
Living with Thalassemia: Challenges and Support
Living with a chronic blood disorder takes a significant physical and emotional toll. The psychological impact of lifelong medical treatments, frequent hospital visits, and managing chronic fatigue can trigger anxiety and depression. Maintaining a high quality of life requires comprehensive care that addresses both physical and mental well-being.
Support groups play an invaluable role for patients and families navigating this condition. Connecting with others who understand the burden of thalassemia provides emotional relief and practical advice. Additionally, patients must remain vigilant about managing long-term complications, scheduling regular cardiac evaluations, monitoring endocrine function (like diabetes and thyroid issues), and staying up-to-date on vaccinations to prevent infections, especially if the spleen has been removed.
Prevention and Genetic Counseling
Because this condition is inherited, prevention mainly focuses on awareness, education, and early screening. Carrier screening is a simple and effective blood test that helps individuals determine whether they carry alpha or beta gene mutations before planning a family. Since carriers often show little or no symptoms, testing plays a key role in identifying hidden risks.
When two carriers decide to have children, genetic counseling becomes extremely important. Genetic counselors help families understand inheritance patterns, possible outcomes for future pregnancies, and available medical options. This guidance allows couples to make informed decisions based on clear medical information rather than uncertainty.
One advanced preventive option is Pre-implantation Genetic Diagnosis (PGD), which is used alongside in vitro fertilization (IVF). In this process, embryos are created in a laboratory and then tested for genetic abnormalities before implantation. Only embryos without the disorder are selected, significantly reducing the risk of passing the condition to the next generation.
Thalassemia vs. Other Blood Disorders
This inherited blood condition is often confused with iron deficiency anemia because both can present with similar signs such as fatigue, pale skin, and small red blood cells. However, the underlying causes are completely different. Iron deficiency anemia results from a lack of iron in the body and usually improves with iron supplements. In contrast, giving iron supplements in this genetic condition can be harmful if iron levels are already normal or elevated, especially in patients who receive frequent transfusions.
Proper diagnosis is therefore essential before starting any treatment, as misinterpretation can lead to unnecessary or even dangerous therapy.
When compared with other inherited blood disorders, it is often discussed alongside sickle cell disease. Both conditions affect hemoglobin, but their mechanisms differ significantly. In this genetic disorder, the body produces insufficient normal hemoglobin, leading to weakened and fragile red blood cells. In sickle cell disease, however, abnormal hemoglobin causes red blood cells to become rigid and crescent-shaped, which can block blood vessels and trigger painful complications as well as organ damage.
Understanding these differences helps improve diagnosis accuracy and ensures patients receive the most appropriate treatment for their specific blood disorder. It is also commonly grouped under different kinds of blood diseases, since it belongs to a broader category of inherited hemoglobin and red blood cell disorders that include several other related conditions.
Research and Future Directions
 The future of treatment for this condition is highly promising, largely driven by rapid progress in genetic science. Gene therapy is emerging as a revolutionary approach, where a patient’s own stem cells are collected, corrected in a laboratory to fix the underlying hemoglobin defect, and then returned to the body to restore healthy red blood cell production. This method reduces or even eliminates the need for donor-based procedures in the future.
The future of treatment for this condition is highly promising, largely driven by rapid progress in genetic science. Gene therapy is emerging as a revolutionary approach, where a patient’s own stem cells are collected, corrected in a laboratory to fix the underlying hemoglobin defect, and then returned to the body to restore healthy red blood cell production. This method reduces or even eliminates the need for donor-based procedures in the future.
In addition, advances in CRISPR gene-editing technology are opening new possibilities for directly targeting and repairing faulty DNA responsible for the disorder. These precise techniques aim to correct mutations at their source, offering the potential for long-term or permanent solutions rather than lifelong symptom management.
Alongside genetic innovations, new drug therapies are being developed to improve red blood cell maturation and reduce ineffective blood cell production. These treatments may help decrease the frequency of transfusions and lessen complications such as iron overload. At the same time, global health programs are working to improve early screening, carrier detection, and access to safe blood transfusion services, especially in regions where the condition is more common.
Navigating the Future with This Condition
Living with this inherited blood disorder involves medical, emotional, and lifestyle challenges. Patients often need long-term monitoring, regular treatments, and careful management of complications such as anemia and iron imbalance. Despite these challenges, modern medicine has significantly improved both life expectancy and quality of life for affected individuals.
With timely diagnosis, proper medical care, and strong family and community support, many people are able to lead productive and fulfilling lives. Genetic counseling and regular screening are especially important for families at risk, as they help in making informed decisions about future pregnancies.
Continued research, better healthcare access, and global awareness efforts are steadily improving outcomes. With ongoing scientific progress, the possibility of more effective and even curative treatments is becoming increasingly realistic in the years ahead.
Conclusion
Thalassemia is a lifelong inherited blood disorder, but with early diagnosis and proper treatment, most patients can manage symptoms effectively. Advances in transfusions, iron chelation therapy, and bone marrow transplantation have greatly improved quality of life. Ongoing research, including gene therapy, offers hope for more permanent cures in the future.
Frequently Asked Questions (FAQ)
1. What is thalassemia?
Thalassemia is an inherited blood disorder that affects the body’s ability to produce normal hemoglobin, leading to anemia and reduced oxygen delivery throughout the body.
2. Is thalassemia genetic?
Yes. Thalassemia is passed from parents to children through inherited gene mutations affecting hemoglobin production.
3. What are the main types of thalassemia?
The two primary types are alpha thalassemia and beta thalassemia. Each type ranges from mild carrier forms to severe disease.
4. What is beta thalassemia trait?
Beta thalassemia trait, also called beta thalassemia minor, occurs when a person inherits one mutated beta-globin gene. It usually causes mild anemia or no symptoms at all.
5. Can thalassemia be cured?
A bone marrow or stem cell transplant is currently the only established cure for severe thalassemia. Gene therapy is also emerging as a promising future treatment.
6. What are the common symptoms of thalassemia?
Common symptoms include fatigue, weakness, pale skin, jaundice, slow growth, enlarged spleen, bone deformities, and severe anemia in more advanced cases.
7. How is thalassemia diagnosed?
Doctors diagnose thalassemia using blood tests such as Complete Blood Count (CBC), hemoglobin electrophoresis, HPLC testing, and genetic testing.
8. Why do thalassemia patients need iron chelation therapy?
Frequent blood transfusions can cause dangerous iron buildup in organs. Iron chelation therapy removes excess iron from the body to prevent heart, liver, and endocrine complications.
9. Can people with thalassemia live normal lives?
Many people with thalassemia can live long and productive lives with proper treatment, regular monitoring, healthy nutrition, and ongoing medical care.
10. Is genetic counseling important for thalassemia?
Yes. Genetic counseling helps carriers and families understand inheritance risks, prenatal testing options, and family planning decisions to reduce the chance of severe thalassemia in children.